
Table of content
- Clinical Biochemistry
- 🐮 Proteins
- 👾 Enzymes
- 🧈 Lipids
- 🍬 DM, Hypoglycemia and Hyperuricemia
- 🍑 Liver, Pancreas, GI exploration
- 🪨 Iron, Heme and Copper Metabolism
- 🦴 Bone, Ca2+, Mg2+, Phosphorus
Member Resources
 Year 4
Year 4 Year 5
Year 5 Year 6
Year 6No access

Clinical Biochemistry
🐮 Proteins
- Proteins are composed of amino acids linked together by peptide bonds.
- Each protein can be described in terms of its structural organization:
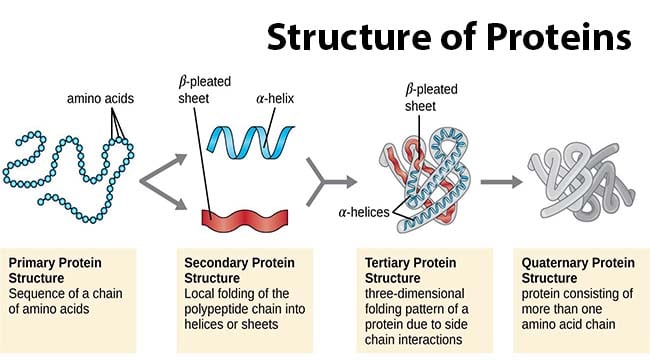
- Primary structure: Refers to the sequence of amino acids in the polypeptide chain, including the N-terminal and C-terminal ends of the molecule.
- Secondary structure: Involves the formation of alpha-helices and beta-sheets, which are local conformations stabilized by hydrogen bonds.
- Tertiary structure: Represents the overall shape of a single protein molecule. It includes protein domains, which are specific structural regions that confer specific functions to the protein, such as enzymatic activity.
- Quaternary structure: Describes the final shape of a protein, which is formed by the assembly of several protein molecules. These individual protein units are often referred to as protein subunits, as seen in the example of hemoglobin with its alpha2beta2 subunits.
Mnemonic: CAT HIPE 🐱
- Complement and coagulation factors
- Acute phase proteins: CRP (C-reactive protein), amyloid protein A, haptoglobin, fibrinogen
- Transport proteins: haptoglobin, transferrin, ceruloplasmin, apolipoproteins
- Hormones (proteohormones): calcitonin, PTH (parathyroid hormone), insulin
- Immunoglobulins: IgG, IgA, IgM, IgE, IgD
- Proteinase inhibitors: a1-antitrypsin, antithrombin III
- Enzymes: amylase, lipase, cholinesterase
Mnemonic: HEI ABC HammerThrow, ps ICH LB DC
- coloidosmotic p. maintainance (mainly albumin)
- source for AA
- Buffer
- Transport
- Iron: hepcidin, transferrin, ferritin
- Calcium: calbindin
- Hormones
- Lipids: apoproteins A, B, C, E
- Bilirubin: albumin
- Drugs: cisplatine uses copper transport channels
- Trace elements: copper - ceruloplasmin
- Enzymes
- Immunity
- Hemostasis
- Heme metabolism
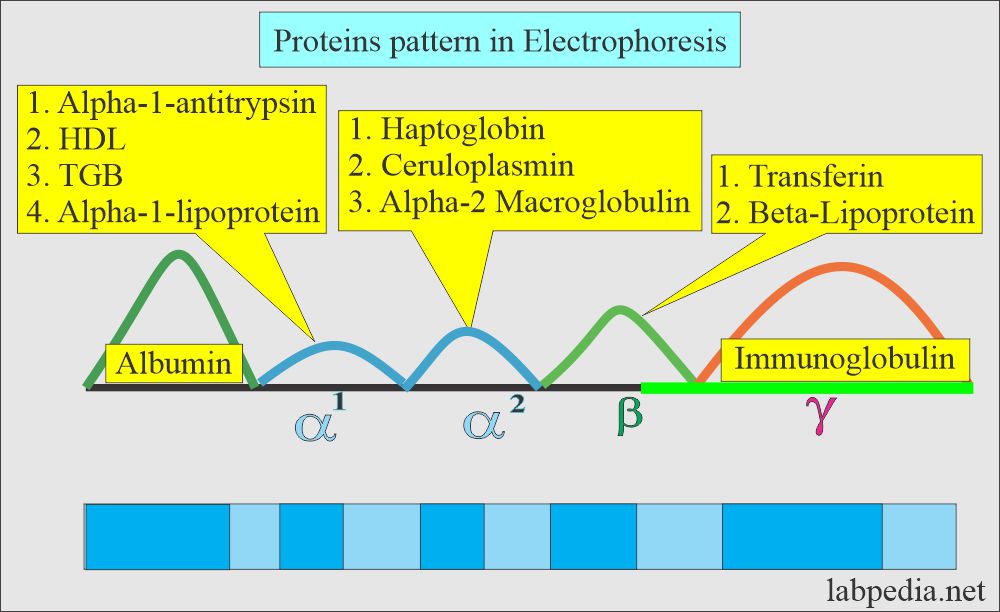
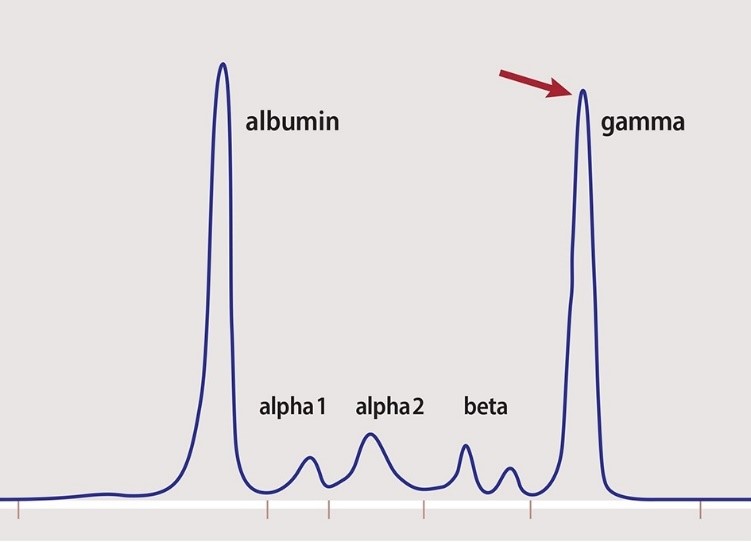
- Is used to separate proteins based on their electrical charge and size
- Used to identify which proteic fraction is modified & to orientate towards a syndrome, NOT the cause
- Based on their movement on an electrical field
- Albumin: 52-60% (1)
- Alpha-1 globulins: 3-5% (5)
- Alpha-2 globulins: 7-9% (4)
- Beta globulins: 11-14% (3)
- Gamma globulins: 16-20% (2)
qualitative + quantitative change in plasma proteins ⇒ change in albumin/globulins ratio [normal>1]
- Normal TPc
- Decr. TPc
- Incr. TPc

- N: With normal total protein concentration:
- Acute phase reaction
- Chronic inflammation
- ↓: With decreased total protein concentration:
- Nephrotic syndrome
- Liver cirrhosis
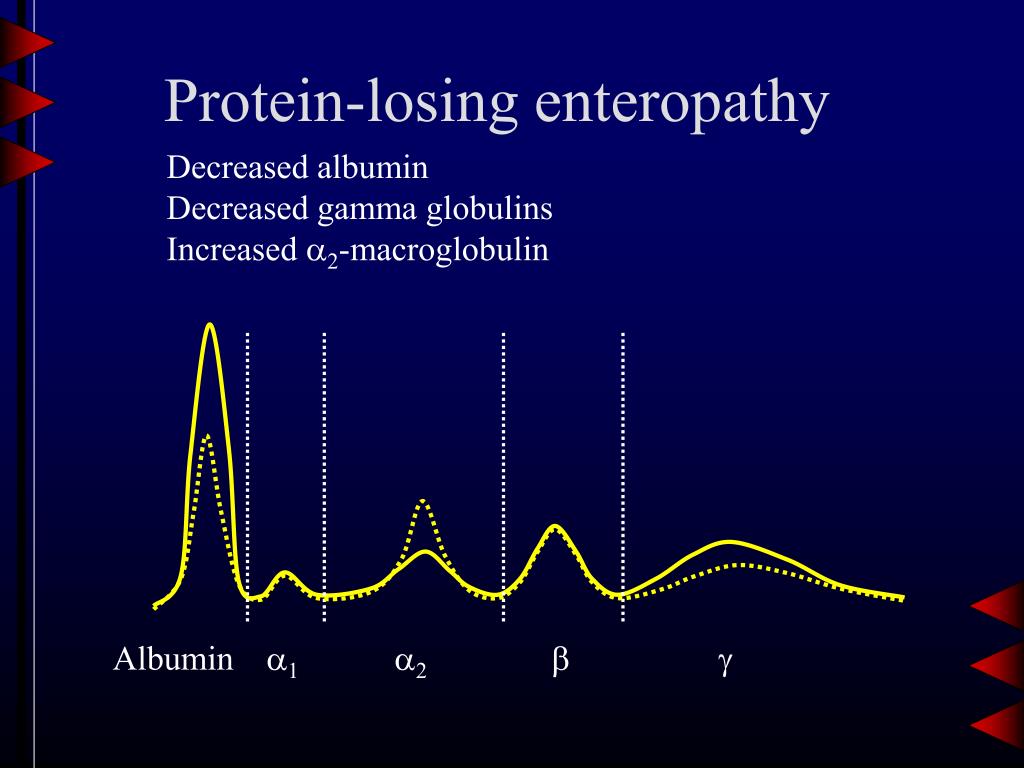
- Exudative enteropathy
- ↑: With increased total protein concentration:
- Multiple myeloma
- Waldenström disease
- Light chains disease
- alpha2 incr. (compensatory)
- albumin decr.
- ↓ albumin (↓synthesis)
- alcohol → IgA incr. ⇒ common block (beta+gamma "bridging")
= blurred distinction betw. beta + gamma
→ sharp gamma peak
only 1 Ig incr. (mainly IgG)

gamma peak (IgM inc.)
- Anemia
- lymphadenopathy
- hyperviscosity syd → internal hemorrhages
kappa + lambda fragments → kidney → amyloidosis
IL1, IL6, TNF
binds to phosphocholine on pathogen → activates complement 📷
- quick incr. CRP
- ESP may be normal in first 24h
- CRP decr. more rapidly after treatment
→ Cardiovascular risk based on CRP levels:
- Low risk: CRP level of 0-2 mg/L
- Average risk: CRP level of 2-4 mg/L
- High risk: CRP level of 4-6 mg/L
- synthesized by the liver
- ↑ in acute + chronic inflammation
- platelet aggregation + clot formation
- avoiding of irritant inhalatin (smoking)
- infusion of A1AT
- liver + lung transplantation
- hereditary (Autosomal dominant)
- secondary: renal failure, nephrotic syd→ loss of ATIII
thrombosis 👣
- recurrent thrombosis + pulmonary embolism in hereditary form
- nephrotic syd → loss of Anti-Thrombin III → higher activity Factor II + X → thrombosis 📷
A tumor marker is a biomarker found in blood, urine, or body tissues that can be elevated by the presence of one or more types of cancer
- detectable in blood when malignant transformation occured
- precise identification of tissue malignancy

- GI
- Liver
- Pancreas
- Breast, Cervic
- CA 15.3
- CEA
- BR27
- EGF
- Cathepsin D


CA125
AFP (alpha-fetoprotein)
- CEA
- bHCG
- SCCA (squamous cell carcinoma antigen)
- cytokeratins (CYFRA)
PSA, free PSA
Calcitonin
- CEA
- CA (19.9)
- NSA (Neuron specific enolase)
- ProGRP
- CYFRA



AFP (alpha feto protein)
CEA
Ferritin
Hypercalcemia (osteolysis)
Alkaline phosphatase
Acid phosphatase
- gonads - testes + ovaries
- hematologic disease
- hepatic
- lymphoma
- melanoma
- neuroblastoma
- CNS tumors
- Neuroblastoma
- Neuroendocrine tumors (small cell lung)
→monitoring treatment of pulmonary+neuroblastoma
Galactosyl Transferase II
Paraproteins = Ig + light chain fragment
Protein M

- ADH
- ACTH
- PTH
👾 Enzymes

- different AA sequence (different structural parts)
- but same catalytic activity (same domain structure)

- active secretion → decr. = insufficient production by cells
- exocrine origin → incr. in obstruction duct or destruction of gland

- intracellular enzymes → incr. into blood due to membrane permeability changes (worst case: necrosis)
GHGDTD
- Genetic defect (muscular dystrophy)
- Hypoxia
- Decrease in glucose transport to the cell
- Distention of tissue (rapid installed hepatomegaly)
- Bacterial toxins or viruses
- Drugs or chemicals
- no. of cells destroyed
- vascularization
- T1/2 of enzyme
enzymes: CK, AST, LDH
non-enzymatic markers: Troponin, Myoglobulin, Albumin, BNP
CK total: 90-180 U/l
CK-MB: <24 U/l
→ If CK-MB not >6% of total CK ⇒ no myocardial destruction
→ In other words: <6% = No MI destruction
- incr. 6h after MI
- maximum 18-29h
- CK-MB might incr. before total CK (B)
- multiple infarcts → usually incr. CK-MB+CKtotal (C)
→ sometimes only CK-MB incr. (D)
repeat CK test after 4h
enzymes are washed away from place of necrosis
- AST-later than CK, less incr.
- LDH- even later + less but longer incr.
incr. more dramatically 📷
measuring the dynamic (before + after, over time)
DD cardiac pain vs chest pain
→ modified by ischemia
- EKG modification before enzyme incr.
- Enzymes → detection secondary MI
- Enzymes in detecting multiple micro infarcts + if hypertrophy or heartblocks
LDH2/LDH1 ratio
- <1 in myocardial infarction
- >1 in healthy individuals
Dermatomyositis → AST, LDH, CK
Progressive muscular dystrophy (Douchenne) → in severe: AST, LDH, CK100UNL;; in carrier woman: CK+aldolase
Polymyositis: → CK
Malignant hyperthermia → CK, AST

- progressive death of muscle tissue → replaced by fibrous tissue → progressive skeletal m. weakness)
- non-inflammatory, no nerve abnormalitiy
- Duchenne → X-linked recessive (mainly affecting boys)
CK + Aldolase
- CK 100x incr. UNL
- AST incr
- LDH incr
diagnostic of risk before clinical signs present
- inflammation of many muscle groups
- CK

give cortisone → better? → myositis
i.e. halothane → trigger Ca efflux of intracellular depositis into myoplasma → sudden + sustained contraction
- metabolic acidosis (incr. lactic acid)
- tachycarda + tachypnea
- muscle rdigitdy
- hyperthermia 41-42°
CK + AST (ASAT)
→ Ca2+ antagonists
reaction pyruvate + lactic acid
→ 📷
- LDH1: heart muscle
- LDH2: red blood cells
- LDH3: lungs
- LDH4: kidney, pancreas, placenta
- LDH5: liver and skeletal muscles
Megalob.: >10x UNL
Hemolytic: >2-5x UNL
→+AST might be slightly incr. in hemolytic
- low reticulocytes
- normochromic, macrocytic (megaloblastic)
- LDH2+3 incr. in leukemia
- VitB12 in biochem
- No response to therapeutic test (VitB12 administration)

- low cholesterol in leukemia → bad prognosis
- Collagen Type 1 → proliferation
- Bone alkaline phosphatase (BAP) → matrix maturation
- Osteocalcin → matrix mineralisation
acid phosphatase
- Osteoporosis (might be decr. or normal)
- Primary + Secondary Hyperparathyroidism
- Rickets / Osteomalacia (VitD defic) → secondary hyperparathyroidism → 2-3x UNL
- Hyperparathyroidism

- Paget diseases

- Osteoblastic tumor (i.e. metastasis from prostate, mamma)
metastatic prolipheration of osteoclasts → osteolysis (acid phosphatase incr)
⇒Osteolytic sarcoma (Ewig, reticulosarcoma)
→ ALP normal
- VitD deficiency
- Hypothyroidism
- EDTA administration after lead intox
- dialysis





- Mama + prostate Ca
- Gaucher disease

🧈 Lipids
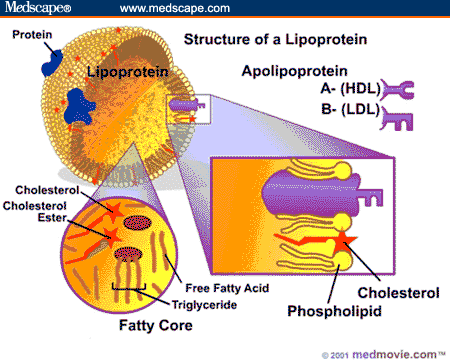
to transport hydrophobic lipids in the blood combined with hydrosoluble proteins
→Role Lipids in general: membranes, VitaminADEK-absorption, hormones,myelin sheath, organ padding
- Biliary acids: emulsification
- Lipase: TG → FFA
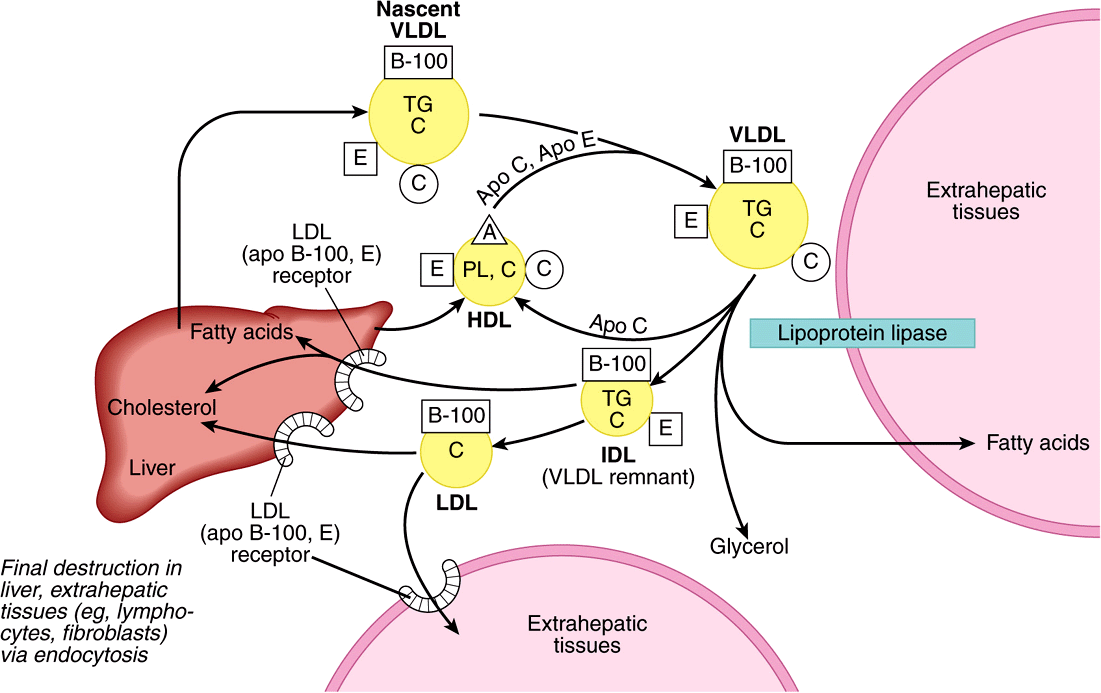
- nascent chylomicrons formation (ApoB) - 80%TG, <15%Cholesterol esters
- Enter blood via lymph → ductus thoracicus
- Receive Apo C II+E from HDL ⇒ mature chylomicrons
- →Tissue → ApoC stimulates LPL → TG into FA,+ Cholesterol → into cells
- Remnant Chylomicron → Liver: ApoE + LDL-R interaction
- TG + CE into VLDL (ApoB-100)
- LPL in peripheral tissue → VLDL into IDL by releasing TG into FA
3.1 IDL → LDL → carries CE to peripheral tissue (has LDL-R(ApoB) on surface) [3/4 ovary, testes + adrenal]
3.2 Part of IDL is directly taken up by the liver
- TG + CE into VLDL (ApoB-100)
- LPL in peripheral tissue → VLDL into IDL by releasing TG into FA
3.1 IDL → LDL → carries CE to peripheral tissue (has LDL-R(ApoB) on surface) [3/4 ovary, testes + adrenal]
3.2 Part of IDL is directly taken up by the liver
Synthesis:
- synthesized by liver + intestine
- liver makes ApoC+E → delivers to intestine
Reverse transport:
- CE in tissue into FC
- uptake by ABCA1-proteins ⇒ nascent HDL
- Plasma: FC → CE by LCAT ⇒ mature HDL-3
→ into liver: Scavenger R B1
→ some of cholesterol esters transferred from HDL-3 to VLDL,IDL,LDL by CETP in exchange for TG ⇒ HDL-2 → to liver + cleaved by hepatic TG lipase → TG cleavage + free Apo A-1
⇒ Reformation of HDL3: ATP-binding cassete transporter A1 (ABCA1) → transfer cholesterol to pooly lipicated particles: pre-HDL or Apo-A1 → HDL3
- uptake Cholesterol from periph. → liver (reverse cholesterol transport)
- repository for ApoE+C for chylomicrons (+VLDL)
- competition with LDL for binding sites on membranes + prevention internalization LDL in arterial wall
- Stimulated Prostacyclin by endothelial cell → prevent thrombus
- Removal of Mp from arterial wall

< 35/45 mg/dl
Implications → see below (hyper/dyslipidemia)
- Diseases associated with abnormal chemistry or metabolism of lipids:
- Obesity
- Atherosclerosis
- Diabetes Mellitus
- Hyperlipoproteinemia
- Fatty liver
- Lipid storage diseases
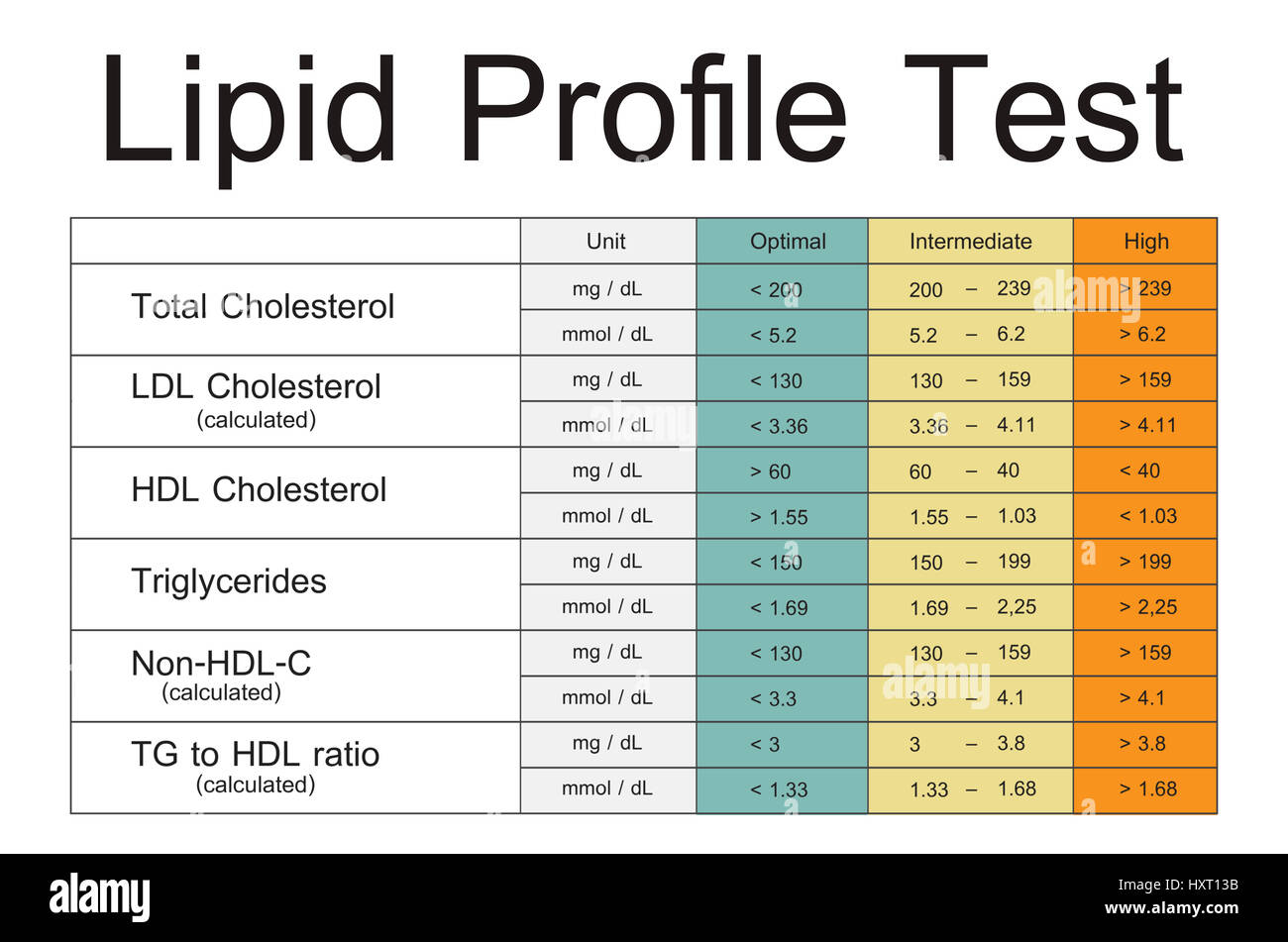
- Reference intervals:
- Total lipids: 500 - 800 mg/dL
- Cholesterol: 110 - 200 mg/dL
- Triglycerides: 50-150 mg/dL
- HDL-chol: >35; >45 mg/dl
- LDL-chol: 70 - 130 mg/dL
HDL3 + HDL2
Synthesis:
- synthesized by liver + intestine
- liver makes ApoC+E → delivers to intestine
Reverse transport:
- CE in tissue into FC
- uptake by ABCA1-proteins ⇒ nascent HDL
- Plasma: FC → CE by LCAT ⇒ mature HDL-3
→ into liver: Scavenger R B1
→ some of cholesterol esters transferred from HDL-3 to VLDL,IDL,LDL by CETP in exchange for TG ⇒ HDL-2 → to liver + cleaved by hepatic TG lipase → TG cleavage + free Apo A-1
⇒ Reformation of HDL3: ATP-binding cassete transporter A1 (ABCA1) → transfer cholesterol to pooly lipicated particles: pre-HDL or Apo-A1 → HDL3
- HDL particles are classified:
- HDL2 (larger, less dense)
- HDL3 (smaller, denser).
- Each HDL particle consists of 4 apolipoproteins.
- HDL can contain apo A-I and apo A-II, or apo A-I alone.
- HDL2 primarily contains apo A-I, while HDL3 contains a combination of apo A-I and apo A-II.
- HDL particles less dense than HDL2 are enriched with apo E.
- DM *
- Hypothyroidism * (↑LDL + ↑IDL)
- Nephritic/Nephrotic (↑VLDL product → ↑LDL)
- Cholestatic liver diseases
- Lp-X
- Lp-Y
- Pregnancy
- Diet rich in fat*
→Abnormal Lipoproteins:





- Alcohol
- Contraceptives oral
- I = familiar LPL def. / ApoCII def.
- IIa = familiar hyperCHOLESTEROLEMIA
- IIb, IV, V = familiar COMBINED hyperlipidemia
- III = Remnant Hyperlipidemia
Type | Fraction modified: |
I | Chylomicrons or LPL |
IIa | LDL |
IIb | LDL & VLDL |
III | Chylomicron Remnants |
IV | VLDL |
V | VLDL & Chylomicrons |
WHO Classification | Cholesterol | Triglycerides |
Type I | < 260 mg/dL | > 1000 mg /dL
(10xUNL) |
Type IIa | > 300 mg/dL | < 150 mg/dl |
Type IIb | < 350 mg/dL | 150-300 mg/dL |
Type III | < 350 mg/dL | 350-500 mg/dL |
Type IV | < 260 mg/dL | 200-1000 mg/dL |
Type V | > 300 mg/dL | > 1000 mg/dL |
Mutation → LDL-R (ApoB100-LDL-Chol-R)
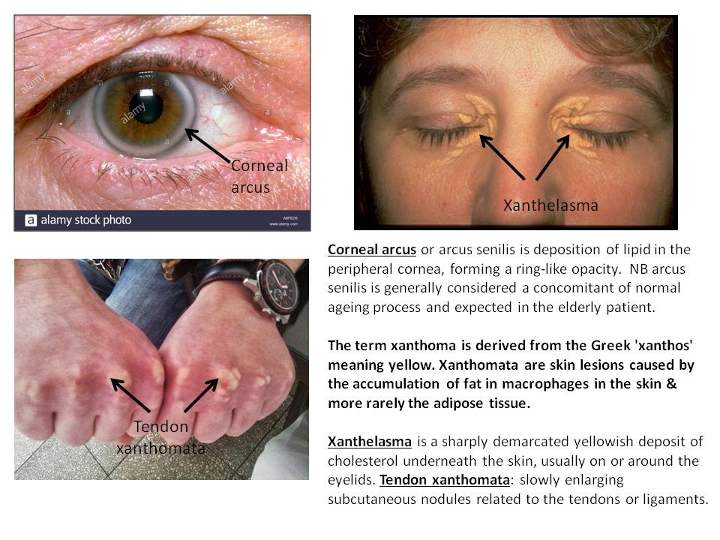
- tendon xanthoma
- xanthelasma
- corneal arcus
CV-D
- Statins
- Niacin
- Bile acid sequestrants
Overproduction of B-100 → VLDL by the liver
no xanthomas!
moderate CV-D risk
- Hypo-alpha-lipoproteinemia (Tangier + LCAT def.)
- Hypo-beta-lipoproteinemia
- A-beta-lipoproteinemia (homozygous)
- Fam. hypo-beta-lipidemia (heterozygot of A-beta)
- Chylomicron retention
- Secondary hypolipoproteinemia

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Mutation in MTTP(microsomal TG transfer prot.) → ApoB deficiency → no CM, VLDL, LDL
- Low TG + Cholesterol (no CM, VLDL, LDL)
- Acantocytes
- Retina degeneration
- Normal at birth, later steatorrhea + growth delay
→ in heterozygots (Fam-hypo-beta): apoB+LDL only ↓50%

- neonates → no intestinal chylomicron production
- LDL + VLDL ↓50%
- chronic cachexia (cancer)
- malabsorption
- Ig-disorders (Ig-lipoprotein-complexes)
- Protein-loosing enteropathy
🍬 DM, Hypoglycemia and Hyperuricemia
- Random >200 mg
- Fasting >126
→ OGGT indicated
- Polydipsiam Polyuria, weightloss
- Glucosuria (if Gluc >180)
- HbA1c >6,3

Measuring C-peptide can help to determine how much of their own natural insulin a person is producing as C-peptide is secreted in equimolar amounts to insulin. C-peptide levels are measured instead of insulin levels because C-peptide can assess a person's own insulin secretion even if they receive insulin injections, and because the liver metabolizes a large and variable amount of insulin secreted into the portal vein but does not metabolise C-peptide, meaning blood C-peptide may be a better measure of portal insulin secretion than insulin itself
Condition | Fasting Glycemia [mg/dl] | OGTT at 2h Glycemia [mg/dl] |
NORMAL | ≤ 100 | < 140 |
DIABETES | ≥ 126 | ≥ 200 |
IGT | < 125 | > 140 |
IFG | 110 – 125 | ≤ 140 |
- DM
- Pancreatitis
- Acromegaly
- Cushings
- Diuretics*
- Glucagonoma
- Pheochromocytoma*
- CRF *
- Starvation
- Insulin overdose
- Insulinoma
- Hypopituitarism
- Addisons
- Hypothyroidism
- impaired renal excretion
- high intake purine-rich food
- high fructose intake

also associated with MS + low dietary zinc
{kind=link}
Allopurinol + Colchicin
🍑 Liver, Pancreas, GI exploration
Infiltration of Plasmocytes
→IgM → primary infection
→IgG → ↑in second. infection + marker of immunity
- Rubella, CMV, Epstein Barr
- Hepatitis
- Alcoholic hepatitis
- Chemicals: Cholorform
- Toxic mushrooms
- yellow fewer
- Gallstones
- Prophyry Cutanea Tarda (see later)
- Chronic Hep - ↑IgG
- Biliary chirrhosis - ↑IgM
- Alcoholic hepatitis - ↑IgA
- Osmotic lysis (excessive water inflow → burst cell membrane)
- Degeneration (disruption of cell membrane
- AST + ALT
- AST mitochondria
- ALT cytoplasms → ↑first (when membrane extens)
- small ↑ in cholestasis
- LDH 5
- Enzymes of the Krebscycle in mitochondria
- OCT (ornithine carbamoyltransferase)
- SDH (succinate dehydrogenase)
- ICDH (isocitrate dehydrogenase)
- GLDH (Glutamat dehydrogenase) in mitchondria
- No + degree of cells damage
- Vascularization of damaged tissue
- Existance of inflammatory barrier
- T1/2 of the enzyme
Non-specific
- Chronic Hep 5-20x
- Compensated cirrhosis 2-3x
- Liver Cirrhosis uncompensated parenchymal normal/slight↑
Esp. AL(A)T
- Acute viral hepatitis 10-50x
Esp. AS(A)T
- Activation of chronic hepatitis → ↑of AST/ALT coeff
- Alcoholic hepatopathy 5-10x
- Tumor - slight ↑
- Acute Necrosis due fungus + organic solvents intox 100x
- biomarker test
- six blood serum values → score ⇒ degree of liverdamage in patient with a variety of liver disease
- same prognostic value as liver biopsy
"Der 2. geborene(Alpha-2-macrogl) haftet (Haptoglobin) am ALTen (ALT) 1.geborenen APOthekter (ALPA1) GeGen (GGT) den er BILIard (Bili total) spielt. Der 2. geborene arbeitet bei der NASA (derivates), er heißt Gilbert und hat 2 acute Probleme (acute hep.+ac. inflamm. syd), die außerhalb der Leber stattfinden (extrahepatic cholestasis + hemolysis=pre+post-hepatic)"
- Alpha2-macroglublin
- Haptoglobin
- Apolipoprotein A1
- GGT
- Bili total
- ALT
"NASA"
- ActiTest - dg. necrotic inflamm. hepatitis
- SteatoTest - dg Steatosis
- NashTest - dg NASH inflamm.
- AshTest - dg. Alcohol liver diseases inflamm.
→likely FP (false pos) or FN (false neg)
→1-5%
- Acute hepatitis
- Acute inflammatory synd.
- Extrahepatic cholestasis
- Severe hemolysis
- Gilbert's synd
→ blood test postboned till feasible
- ALP
- biliary acids
- GGT- ↑alcohol, rifampicin, etc.
- 5Nt (nucleotidasis) - doesnt ↑!
- Urinary bilirubin - indicate ↑direct bili
Causes for cholestasis
- Obstruction
- Hepatocellular damage
- Liver steatosis (in metabolic snd)
- Cholinesterase
- ↓ → ↓hepatic proteinsynth
- anemia
- malnutrition + -absorption
- acute phase reaction
- organic solvent intox
- ↑
- obesity
- HyperLP - IIa, IV, V
- nephritic (acutally nephrotic??)
- PT (QuickT)
- acute liver insuff → pentru ca FVII short T1/2
- Albumin
- liver chirrhosis → shift in ascitis fluid
- not useful in acute
Hematological
- Target cells (hepatic disease)
- megalocytes (alcohol)
- PT- oral anticoagulant (warfarin)
- aPTT - i.v. anticoagulant (heparin)
Antibody Titers
- Mitochondrial Ab - primary biliary cirrhosis
- Antinuclear factor + Smooth muscle Ab - Autoimmune liver+biliary tree
Viral Hepatitis Ab + Ag
Hepatitis C
- HCV Ab
- C Viremia (DNA??)
Hepatitis A
- HAV Ab
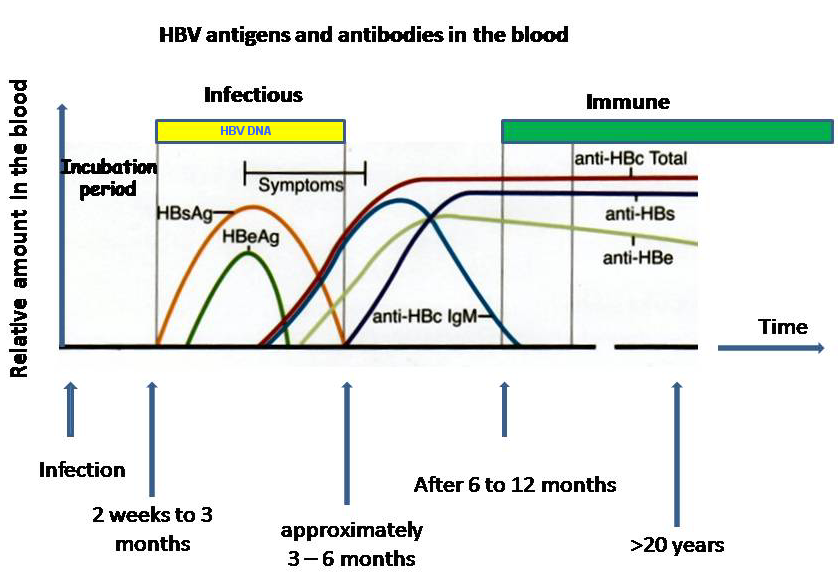
Hepatitis B: 📷
{kind=link}
- HBsAg - acute infection / chronic infection w/ unresolved antigenemia
- HBsAb - recovery + immunity, when Ag disappears, Vaccination→check titer every 5 years
- HbeAg - before onset of symptoms, after HbsAg, active replication + most infectivity
- HBeAb - resolving / no complicating liver diseases, after Ag disappears, pos. in asymptomatic carriers
- HBcAb - after HbsAg disappears, marker for immunity throughout life
- Wilson D
- ↓Copper (serum + urine)
- ↓Ceruloplasmin
- Hemochromatosis
- ↑Iron + Ferritin
- >50% Transferrin saturation
- Alpha-1 antitrypsin deficiency
- ↓ in serum
- Primary liver cell cancer
- Alpha fetoprotein
- mild ↑in hepatitis + alcoholic cirrhosis
JAUNDICE = total bili >3 mg/dl
Prehepatic Jaundice
- ↑unconj. bili >0.5mg/dl
- Hemolysis
- thalassemia
- transfusion w/ incompatible blood groups
Hepatocellular jaundice
→deficit of uptake, conjugation or secretion of bili
→ generalized hepatic dysfunction, also ↑AST+↑ALT
- Infections
- Chemicals, drugs → alcohol, paracetamol
- Genetic → Gilbert, Cringler-Najjar, Rotor, Dubin-Johnson disease, Wilson D, alpha1 antitrypsin
- Autoimmune hepatitis
Post-hepatic jaundice
→ ↑conj. in blood → pale stools + dark urine
- Intrahepatic bile ducts → primary biliary cirrhosis, cholangitis
- Extrahepatic bile ducts → stones, pancreatic tumor
Laboratory/Test | Prehepatic Jaundice | Intrahepatic Jaundice | Extrahepatic Jaundice |
Stool Color | Darker | Light, clay-toned | Pale, clay-like |
Indirect Bilirubin | Increased markedly | Increased | Normal |
Direct Bilirubin | Within normal limits | Increased | Increased markedly |
Urinary Bilirubin | Within normal limits | Increased | Increased markedly |
Urinary Urobilinogen | Increased markedly | Normal or slightly elevated | Decreased |
Urine Color | Dark in hemoglobinuria cases | Dark | Very dark |
Cholestatic Enzymes | Within normal limits | Increased | Increased markedly |
Transaminases | Within normal limits | Increased | Normal |
Ultrasound | Normal bile ducts, hepatomegaly may be present in hemolytic diseases | Bile duct dilation, potential double-duct sign | Dilated bile ducts, potential double-duct sign |
- esp. in prematures
- first 10days of life
short lifespan of fetal hemoglobin → ↑unconj. bilirubin → insufficient conjugation due to immature conjugation enzyme (UDP-glucuronyl transferase) → unconj. bili passes BBB (hydrophbic) → nuclear jaundice (kernicterus)
- UV phototherapy → transforms into hydrosoluble bili → exc. in urine
- Phenobarbital → give preventive to mothers with early birth risk → cross placenta → induces UDP glucoronyl transferase synthesis
⇒ if unsolved after 10day → pathological?!
- Alcoholism
- Biliary tract disease
- precipitated by: ↑Chylomicrons (TG>400mg/dl)
- ↑Amylase serum → 5-10x UNL
- ↑Amylase urine
- ↑Trypsin serum
- ↑Lipase urine+serum
- ↑Glucose
- ↑TG
- ↑Bili
- ↑Renal markers - Crea+Urea
- ↓Ca2+
- due to acute phase reaction → ↓albumin → ↓Ca
- Hydrolysis of Fat by Lipase → insoluble soaps
- ↑first 6h after onset
- remain ↑ for 2days
- remain longer in acute kidney insuff
- Gastric acidity: Basal + stimulated
- ↑pH - pernicious anemia
- ↓pH - Zollinger ellison syd
- observed in treatment of ulcers
- verfication vagotomy in ulcer treatment
- H. Pylori
- Urease breath test
- H.Pylori Ag
- H.Pylori Ab
- Biliary drainage
- macroscopic → tubrid, purulent?
- microscopic → leukos, epithalial cells, cholesterol crystals, tumor cells, giardia cysts
- Biliculture
- in purulent cholecystitis
- Occult Blood test (Gregerson) → detect heme
- Complete Hemogram → in GI hemorrhage (normochromic + -cytic anemia)
- Microscopy → muscle fibers, lipid droplets, starch granules
- Coproparasitologic examination → parasites + eggs
- Copro(stool)culture → Shigella, Salmonella, E.coli
🪨 Iron, Heme and Copper Metabolism
- Hemoglobin
- Myoglobin
- binds Oxygen quicker than Hb
- ↑physical effort
- ↑muscular lesions (rhabdomyolysis)
- Heme containing enzymes in mitochondria involved in cellular respiration
- cytochromes
- cytochromoxidase
- Transferrin (Fe+Siderofilin)→ binds Fe3+
- Siderofilin → beta globulin, contain 1-3 Fe3+ molecules
- Receptors in Liver, Placenta, Erytroblasts+Reticulocytes
- ↑R in Iron def.
- ↓R in Iron overload + ↑Apoferritin (→storage)
{kind=link}
- Fe3+ (non-heminic form) → Fe2+ (heminic form)
- active transport into mucosal cell
- can be stored here → as Ferritin (Fe3+)
- or as Fe2+ through Ferroportion out of the cell
→ conversion to Fe3+ + binding to Transferrin
- ↓ by Phosphates (milk products) + oxalates (green vegetables)
- ↑Vit C → Fe3+→Fe2+
- To Mitochondria → hemesynthesis
- Storage → 60% Ferritin(hydrosoluble) + 30% Hemosiderin (insoluble)
- Liver, Spleen, Bonemarrow → each get 30%
- ↓Ferritin in iron def. anemia (most sensitive marker)
- ↑Ferritin in hepatic inflammation, overload
- ↑Hemosiderin in iron overload (Hemochromatosis →prusian blue)
- central regulator of iron metabolism
- synthesis by liver
- binds feroportion → internalization + degradation → ↓iron efflux from enterocytes + macrophages into plasma & also ↓duodenum absorption
{kind=link}
- Normal homeostasis → ↑plasma iron → ↑hepcidin → iron consumed → ↓hepcidin
- Inflammation → IL6 + other cytokines → ↑hepcidin
- Hereditary Hemochromatosis
→ due to Hepcidin deficiency → mutation genes for hepcidin / hepcidin regulators
→ due to Hepcidin resistance → mutation ferroportion
⇒excessive absorption + transferrin saturation → accumulation of non-transferrin-bound iron in various organs
- Iron in serum + urine
- Serum: Iron bound to Transferrin
- Urine: for iron overload dg., ↑after deferoxamin administration
- ↑Hemolytic anemia, iron overload
- ↓iron def., chronic inflammation, menstruation
- Ferritin
- most sensitive for iron def. anemia (early ↑)
- ↑Leukemia + talasemia
- Transferrin
- ↑small children + pregnancy
- IBC (iron binding capacity)
- =max. conc. of iron transferrin can bind
- ↑ - iron def, hemorrhage
- ↓- iron overload, hepatic disease
- false ↑: chloramphenicol
- blood loss → menstrual, ulcer, tumor
- inadequate diet
- inadequate absorption → stomach or small intestine resection, chronic diarrhea, malabsorption syd, achlorhydria (↓HCl)
- congenital atransferrinemia **→ hypochromic, microcytic anemia; ↓transferrin; resistance to iron therapy; treatment: purified tranferrin
- ↓ - Ferritin, Iron, Hb, Reticulocytes
- ↑ - IBC
- Blood smear: normal/hypochromic, microcytic, anisocytosis (advanced stages)
- deficiency of mitochindrial alpha-glycerophosphate oxidase → lactic acidosis (muscular fatigue)
- MAO deficiency → irritability
- no ↑ of thyroid hormones in response to cold → disturbed thermogenesis
- Atrophy of digestive mucosa → lingual, pharyngeal, esophageal
- Sideropenic dysphagia → Burn sensation + difficulties swallowing
- Hemosiderosis - after blood transfusion → iron in macrophages, no tissue injury
- Hereditary hemochromatosis
- deposition of iron in liver, heart, kidney → organ failure
- ↑Iron + Transferrin saturation→↓IBC
- esp. in males
- mutation of p.C282Y ("Am Pc sitzt Noa mit ner Fahne, but Why?") → Abnormal HFE protein → hepcidin deficiency + ↓interaction Transferrin + Transferrin-R
- Acquired hemochromatosis → due to acute event of talassemia, lead poisoning, chronic excessive absorption
- Sideroblastic anemia → iron overload mitochondria → repression D-ALA → ↓heme
- Aceruloplasminemia → Mutation Ceruloplasmin gene → accumulation iron in pancreas, liver, brain
{kind=link}
- subgroup of neurological porphyrias
- Trias: Abdominal pain, neuropsychiatric +neuromuscular abnormalities (peripheral neuritis, resp. paralysis)
- porphobilinogen deaminase deficiency (usually porphobilinogen → uroporphyrinogen III) → ↑ALA-synthetase acitivity → ↑porphobilinogen + ↑ALA in urine (prophyrin precursor)
- Urine exposed to light → becomes darker
- !no skin lesions → only porphyrin precursors accumulate, not porphyrins
- deficiency uroporphyrinogen decarboxcylase (in porphyria cutanea tarda)
- accumulation of porphyrins in skin → light exposure → ROS → photosensitivity + skin lesions (blisters, edema, necrosis, pigmentation)
- normal urinary ALA + porphobilinigen
- ↑urinary uroporphyrin
- 3 subtypes of cutaneous porphyrias
- Porphyria cutanea tarda - most common, due to liver disease or excessive alcohol intake → ↑uroporphyrin
- Congenital erythropoietic porphyria (↑uroporphyrinogen + ↑coproporphyrinogen)
- (Erytropoetic) Protoporphyria (↑protoporphyrin)
- inhibits ALA-Dehydratase + Ferrochelatase
- ↓Porphobilinogen (PBG)
- Complications:
- Lead line (gingiva)
- Encephalopathy
- Anemia (decr. erythropoesis)
- Basophilic strippling of RBC
- Intestinal colic
- Wrist + Foot drop (periph. neuropathy)
- Renal tubular acidosis
Autosomal recessive mutations in the ATP7B gene (Wilson gene) on chromosome 13
- Encodes for a membrane-bound, copper-transporting ATPase
→ Defective ATP7B protein:
- Reduced incorporation of copper into apoceruloplasmin
- Reduced biliary copper excretion
- Accumulation of ↑ free copper in the liver, cornea, CNS (basal ganglia, brain stem, cerebellum), and kidneys
Organ System | Corresponding Symptoms |
Liver | Acute liver failure, chronic hepatitis, cirrhosis, hepatosplenomegaly, portal hypertension, abdominal pain, jaundice, ascites, hepatic encephalopathy |
Neurological | Motor disturbances (dystonia, parkinsonism, tremor, wing-beating tremor), dysarthria, behavioral changes (depression, irritability, psychosis), cognitive impairment |
Renal | Fanconi syndrome |
Eyes | Kayser-Fleischer rings |
- Mutation ATP7A gene (in wilsons ints 7B) - x-linked-recessive
→ accumulation in small intestine + kidney
→ poor distribution of copper (↓↓brain, bone, skin, hair, bloodvessels)
→seizures, split inner walls of brain arteries → rupture or blockage, subnormal bodyTemp, neurodegeneration in gray matter
→ osteoporosis
→ colorless fragile hair
- Screening: urinary homovanillic acid/vanillylmandelic acid ratio
- Treatment: iv/sc copper
🦴 Bone, Ca2+, Mg2+, Phosphorus
- tp1NP - Matrix prolif. → collagen type 1 synth.
- BAP (bone alkaline phosphatase)- Matrix maturation
- Osteocalcin - Matrix mineralization
"Der Wasser-Pro ist in der line (Hydroxyproline) vor der pyramide (pyridinoline) im schneidersitz (beta cross lap aka CTX) mit seinem Teleskop (C+N Telopeptide)
- Hydroxyproline (urine)
- Pyridinoline + Deoxypyridinoline
- C Telopeptide - CTX (beta cross lap) 📷
- N Telopeptide (urine + serum)
- Urinary calcium (not used anymore)



no → dg by DEXA (dual energy xray absorptiometry)
⇒but used to check therapy response (biochem markers after max 3month, DEXA 1 year)
MC PTSD bei der DB hat Nierensteine
- primary hPTH (adenoma)
- Hyperthyroidism
- VitD-intox (prolonged administration)
- Bone tumor (primary or mets)
- Diuretics (Thiazides)
- Sarcoidosis (prod. of calcitriol in Mp + cells of granulomas)

- Milkalkali synd. (ingestion calicum+absorbable alkali (milk, calcium carbonate)

- Calcinosis cutis

- HypoPTH (post adenoma surgery)
- PseudohypoPTH (kidneys dont respond to PTH)
- VitD deficiency (Rickets, Osteomalcia)
- Hypersecretion Calcitonin (thyroid tumor)
- CRF (decr. VitD + Urea fixate Ca to unionisable form)
- Acute Pancreatitis (Ca-FattyAcid-Soaps)
- Laxatives* (Mg+P)
- Loop-Diuretics*
- VitD resistant rickets - alpha1 hydroxylase deficiency
DAD aufm RADT fällt(hypothyroid)
- RF (cant excrete)
- Diabetic acidosis (High excretion of H+ and K+ → high Mg reabsorption + incr. secretion into the blood Mg together with K)
- Addisons *
- Dehydration *
- Hypothyroidism
- Antacids with Mg



- Diuretics (K-sparing [Thiazide - ↑calcium-induced magnesium absorption [but acutally bullshit])
Increase
- Hemolysis (most Mg in RBC)
- Kidney lesions in prolonged salicilates + lithium treatment
Decrease
- Drugs: Aldosteron, Insulin, Diuretics with mercury
- Calciumgluconate - interference with testing methods
- alcohol +(incr. secretion by directly acting on kidney)
- chronic diarrhea +
- diuretics (Loops) +
- hemodialysis (removal)+
- CKD (decr. reabsorption??)+
- cirrhosis (low dietary uptake, greater urinary secretion)+
- chronic pancreatitis(malabsorption)+ + malabsorption
- hyperthyroidism + hypoparathyroidism

- prolonged lactation +
"Das aggro(acromeg) kid (kidney insuff) mit dem riesen boner dick (bone tumor+VitD) hat keinen hypoparathyroidgland. Deswegen geht er zu Rick ins krankenhaus der im Coma liegt weil er zu viel insulin gespritzt hat und klaut ihm seinen Hyperparathyroid gland."
- kidney insuff (cant be excreted)
- hypoparathyroidism (no stimulation of urinary excretion via PTH)
- ↑VitD (incr. reabsorption)
- Acromegaly (incr. reabsorption due to GH stimulation=
- bone tumor (incr. bone loss)
- Hyperparathyroidism (incr. excretion)
- Rickets (decr. VitD activity on reabsorption)
- Diabetic coma (treatment??→see below)
- Hyperinsulinemia (Stimulation of glycolysis → ↑formation of phosphorylated carbohydrate compounds in liver+skeletal muscle)