





Hematology
6 month - 5 yrs Hb < 11 g/dl
5-11 yrs Hb < 11.5 g/dl
12-14 yrs Hb < 12 g/dl
>15 yrs females Hb <12 g/dl
>15 yrs male Hb < 13 g/dl
Via the MCV (mean corpuscleral volume)
- < 80 Microcytic
- 80-100 Normocytic
- >100 Macrocytic

*HUS
Hemolytic-Uremic-Syndrome (HUS)
- iron
- iron deficency (normal at onste tho!)
- sideroblastic anemia ( Lead poisoning)
- Thalassemia
- chronic disease ( normal at the beginning as well)

- megaloblastic anemia
- Macro or Normo
- Loss anemia:
- hemolytic anemia
- acute blood loss
- BM failure
- aplastic anemia
- bone marrow infiltration
- chronic disease and iron deficency at the onset
- Macro or normo
- Loss anemia:
- hemolytic anemia
- acute blood loss
- BM failure
- aplastic anemia
- bone marrow infiltration
anamnesis:
- fatigue
- palpitations
- dyspnea
- activty intolerance

- pale skin
- new systolic murmur + taxhycardia (compensatory with high flow murmur)
- spoon shapes nails
- angular chelitis of the mouth
- atrophic glossitis
Mean corpuscular volume (MCV)
it allows you to judge, if the rbcs are too small, normal or too big,
so you can say macro-/ normo and microcytic
is the variation of volume of the RBCs
are immature blood cells, that circulate, are slightly larger than normal rbcs → high number can alter the MCV
- Transport protein for iron
- Can be reported as total iron binding capacity (TIBC)

main storage protein for iron (so if low, there is no iron storage)
BUUUT its an acute phase reaction protein as well
- Measurement of circulating iron most of which is bound to transferrin
- Serum iron levels are subject to physiological daily fluctuation and can be influenced by dietary intake. → not so informative

↓ lifespan of RBCs due to increased destruction
- intravascular ( in the circulation)
- extravascular (in liver or spleen)
120 days
it means there is a problem within the hem metabolism, leading to accumulation of iron in the mitochondria → has ring sideroblasts as a consequence
- 🍑 🍠hepatomgealy and splenomegaly (coz they be workin to distroy them RBCs and reticuloendothelial hyperplasia)
LAB :
- ↑ Reticuolcytes
- ↑ unconjugated bili +LDH

- ↑ urinary urobilinogen
- positive Coombs test (antoglobulin test)→ in immun mediated hemolysis, you can check for the antibodies
- weird shape of the erys (surprise)
the dont look doughnut shaped like usual, but sphere-shaped (wow) 📷
its a heterogenous autosomal dominant hereditary disorder that affects the structural proteins (ankyrin the most often) of the RBC membranes and cytoskeleton → spherocytes as a consequence
F highly variable, from completely asymptomatoc to severe symptomes
very early gallstones because of increased bilirubin excretion!
- usually mild symptomes
- mild spleonmegaly
- mild anemia
- sometimes jaundice
- normocytic anemia
- MCHC normal
- RDW increased
- hemolysis marker increased (LDH, bili, urinary UBG…)

{kind=link}
{kind=link}
osmotic fragility test
due to the instable cytoskeleton the spherocytes lyse easier than concave RBS → exposure to hypotonic saline solution makes them swell and hemolysis happens
but its not very sensitive and not specific to HS
autoimmue or isoimmune ABO incompatiblity in neonates
- typical features
- splenomegaly
- spherocytes
- reticulocytosis
- negative coombs test
- (family history)
folic acid supplementation
- 🍠Splenectomy (after yr 6)
- vaccinations are importaint before!
- lifelong oral penicillin prophylaxis
- 🌝Cortisteroids to control severe crisis
- 🆎 blood transfusion <5
- cholesystectomie if gallstones are symptomatic


disorders, that affect the structure and function or snynthesis in hemoglobin
HbF, HbA and HbA2
HbF has a greater oxygen affinity
adults dont have fetal hemoglobin anymore
2 pairs of globin chains, 4 heme groups, consiting of iron and porphyrin

- Globin disorders
- qualitative ( eg Sickle cell)
- quantitative (eg Thalasemia)
- HEME disorders (carboxyhemoglobin)
a deficient globin chain synthesis, resulting in an imbalance between alpha and beta globin chain prodution (reduced beta chain production)
autosomal recessive, mutation of the beta gene for hemogloblin chain
- mediterranian area
- africa
- southeastern asia
apparentlyyy it is (T)
- major
- intermedia
- minor
- 📈failure to thrive
- 🐹“chipmunk face” 📷
- maxillary overgrowth
- frontal bossing
- exposed front teeth
- 💪Skin changes
- pallor
- jaundice
- hemosiderosis 📷
- 🦴bone problems
- osteoporosis
- fractures
- hair on end sign in x ray of skull
- pulmonary hypertension
- hypersplenism, hepatomegalie
- chirrhosis
- endocine problems
- hypothyreodism
- diabetes mellitus

- low hb
- increases RDW
- low MCV
- hypocromic, microcytic rbcs
- 🎯 target cells 📷
hemoglobin electrophoresis
- mild forms dont need treatment
- blood transfusions in hb< 7
→ every 3-5 weeks
- spelenctomy
- treatment of other problems like osteoporosis, gallstones, endocrine deficencies
manifest hemoglobiopathinopathie associated with hemoglobin S and sickle cells
autosomal recessive, HBB gene mutation, point mutation, glutamine changed to valine
along the Malaria areas 📷, as it is a protective factor against malaria
{kind=link}
- painful vaso occlusive crisis (micro thrombs due to sickle cells) → in any organ of the body and are manifested by pain and/or significant dysfunction
- 🫀acute chest syndrome
- 🧠stroke
- 🍑intrahepatic crisis
- 🥐kidney infarctions
- 🍆Priapism
- Hemolytic crisis (severe anemia)
- ⇒ 🍠acute splenic sequestration ⇒ 🧫 increased risk for infections

→ all results of infarctions due to the sickle cells!
- failure to thrive
- 💪skin ulcers (bcs of infarction)
- 🦴avascular, aseptic necrosis of bones
- 🫁pulmonary fibrosis, asthma
- 🫀cardio:
- murmurs, cardiomegaly
- congestive heart failure
- pulmonary hypertension
- 🍑hepatomegaly
- 🍠splenomegaly
- 🦠transfusion related disease
- hepatitis
- hiv
- 👁️proliferative retinopathy
- 🥐Kidney:
- hyposthenuria/isosthenuria
- proteinuria, nephrotic syndrome,
- chronic renal failure, hematuria (papillary necrosis)
- sickle cells
- target cells
- howell jolly bodies
is 👎
median life expectancy 42-58 yrs
can cause premature death in children
- non pharma: 🛏️bed rest
- 🆎RBC transfusion
- 💥symptomatic
- analgesia
- hydration
- 🧫AB for infections
- 🦟 Hydroxycarbamide (hydroxyurea) increases HbF production!
- 🍠 + 🧫
- immunization (vaccination)
- daily penicillin
- folic acid supplementation
- trigger avaoidance
- :BM transplant in young patients
8-10mg
Vitamin C
proteins
- ↓ intake
- generalized malabsorbtion
- celiac disease, Inflammatory bowl disease
- gastritis /high pH
- genetic
- poor vit c intake
- Iron chelators
- GI bleeding
- ulcer
- diverticel
- Menstruation
- growth
- EPO treatment
- cyanotic congenital heart disease (polycytemia)
- pallor
- tiredness
- tachycardia
- PICA

- Brittle nails
- koilonychia (spoon-like nail deformity)
- hair loss
- altered behavior, intellectual and motor function (always reversible)
microcytic hypochromic anemia
- classic presentation. microcytic hypochromic anemia (not necessarily)
- low Hb
- microcytosis
- hypochromia (↓MCH)
- low/ normal MCHC
- ↓transferrin saturation (↑TIBC)
- Hb response to oral iron is the most reliable criterion of iron-deficiency anemia
- wide variations
- decreased by infection
- increased by liver disease, hypoxemia
⇒ see diagnostic algorithm ⇒ DDx with microcytic anemias
Parameter | Iron deficiency anemia | β-Thalassemia minor | Anemia of chronic inflammation | Sideroblastic anemia |
Prevalence | ++++ 🥇 | +++ 🥈 | ++ 🥉 | + (rare) |
MCV | ↓ | ↓↓ | Normal / ↓ | ↓ / Normal / ↑ |
Serum iron | ↓ | Normal / ↑ | Normal / ↓ | Normal / ↑ |
TIBC or transferrin | ↑ | Normal / ↓ | Normal / ↓ | Normal / ↓ |
Transferrin saturation | ↓ | Normal / ↑ | Normal / ↓ | Normal / ↓ |
Serum ferritin | ↓ | Normal / ↑ | Normal / ↑ | Normal / ↑ |
Bone marrow reticulo-endothelial iron stores | ↓ / absent | Normal / ↑ | Normal / ↑ | ↑ |
Marrow sideroblasts | ↓ / absent | Normal / ↑ | Normal / ↑ | Ringed sideroblasts |
Reduced marrow iron stores → ↓Ferritin → ↑Transferrin/TIBC → ↓ Transferrin saturation → ↓Serum iron → ↑FEP (Free erythrocyte protoporphyrin)→ ↓MCV → ↓ Hb
check regularly, dont drink cow milk before 1 yr old, if necessary iron supplementation
- correct the cause (blood loss/ low intake?)
then:
- dietary advice
- supplementation
- blood transfuison (BUT TRY TO AVOID IT! should almost never be recommended for dietary iron deficiency, even in children with an Hb as low as 4 g/dL)
12-24hrs subjective improvement
Increase in Ferritin, takes 2-5 month!
it makes abdomnial pain, black stools and constipation!
TTTTT 30% of all cancers
Malignant | Inflammatory | |
Size | >2cm | <2cm |
Surface | firm + irregular | rubbery |
Tenderness | ❌ | ✅ |
Mobility | ❌ | ✅ |
multiple sides | ✅ | ❌ |
B-symptoms | ✅ | ❌ |
- Acute
- 80%: Acute lymphoblastic leukemia (ALL)
- B lymphoblastic 80%
- T lymphoblastic 14%
- Burkitt 1%
- 20: Acute myeloblastic leukemia (AML)
- Chronic (rare)
⇒ 📷: myeloid + lympoid
{kind=link}
Myeloid or lymphoid progenitors (aka precursor “BLASTS”) cant differentiate
→ buildup of blast cells (normal 1-2%; acute leukemia >20%) in BM ⇒ CROWDS OUT ALL BLOOD CELLS in BM: RBC, Plts, Leukos → anemia, bleeding, infections
⇒ secondary involvement of retculoendothelial system (LN + HSM) + other organs (bones, joints, CNS, testes, skin)
≥20% Blasts in BM (leukemia)
⇒ Lymphoblasts! (lymphoblastic)
- 💣sudden onset (d-w)
- BM-failure
- → 🐼🩸🧫signs of anemia, neutropenia + thrombocytopenia
- →🦴 bone/joint pain
- 🅱️-symptoms
- 🔀Infiltration
- 🟢Lymphadenopathy (cervical / mediastinal)
- 🍑🍠HSM (hepatosplenomegaly)
- often at relapse
- 🧠CNS (mass effect)
- 🥜Testes (enlargement)

All metastatses in CNS or testes !
- CBC: → anemia + thrombocytopenia
- Periph smear: Blasts!
- Bone marrow: blast >20%
- Immunophenotyping:
- Lymphoid markers for B (CD10, 19,20) and T (CD 2-8)+ early lympoid markers
- Myeloids markers for Granulocytic (CD13, CD33)
- prognostic testing
- Karyotyping (cytogenic analysis)
- normal → good prognosis
- translocations (esp. philadelphia + t(4,11) → bad prognosis)
- Molecular testing (PCR, sequencing)
⇒ variable WBC!!
→ FLT3 mutation(bad prog)
- Pancytopenia ⇒ AML, aplastic anemia
- Lymphadenopathy + splenomegaly ⇒ EBV/ lymphoma
- bone/joint pain → JIA (juvenile idipath. arthritis)

Early and aggressive chemotherapy improves the prognosis, e.g., 80–90% of patients with ALL achieve complete remission with chemotherapy
- Remission induction + CNS protection 4 weeks
- Consolidation + CNS protection 4 weeks
- Maintenance 2-3 years
- Intensification (during maintenance) 8 weeks

- SHORT TERM
- BM + immunosuppression 🐼🧫🩸
- ; 🤮Anorexia +💇♀️ Alopecia
- LONG TERM:
- 2️⃣secondary malignancies
- 🥜infertility
- 🧠Neuro + ❤️cardiac impairment
- 📈↓growth
(anemia, infections, bleeding)
bimodal
15-34y AND >50y
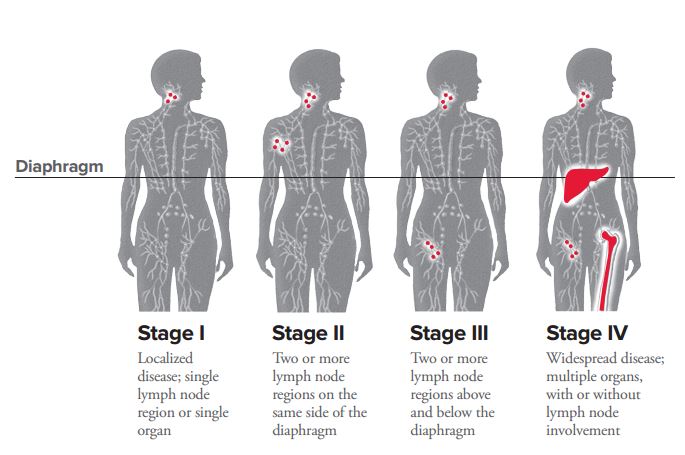
⇒ both B-symptoms (less common in NHL)
- HL:
- 🍏Lymphadenopathy:
- cervical
- supraclavicular
- mediastinal
- 🍑🍠HSM (when liver/spleen involv)
- rare, but specific: 🍷pain after alcohol,🔥 Pel-Ebstein fever, (pruritus)
- NHL:
- rare nodal involvement → when involvement: cervical
- 🫀Mediastinal lymphoma ⇒ compression (SVC-syndrome + airway)
- 💩Abdominal lymphoma ⇒ intussiception, ascitis
Pel-Ebstein fever:


→ biopsy (lymphnode, bonemarrow, liver):
⭐️Reed-Sternberg + 🟢Hodgkin cells + reactive normal cells
→ chest + LN imaging📽 xray, CT, PET, US → CSF in NHL
→Lab:
- High LDH🤥
- high ESR + anemia🐼
- Leuco incr or decr (Neutrophilia, Eosinophilia 🥀+ Lymphopenia)
- Altered liver test🍑 (ALT, AST, bili)
{kind=link}
{kind=link}
🔬HISTOLOGY → outcome depends on lymphoma-type
Non-Hodgkin Lymphoma (NHL) - 60% Subtypes:
a. Small Noncleaved (Burkitt) Lymphoma - Mature B-cell
- (1) Sporadic (Europe, North America)
- (2) Endemic (Africa) - Strong association with Epstein-Barr virus (EBV) infection
b. Lymphoblastic Lymphoma - T-cell (Rarely pre-B-cell)
c. Large Cell Lymphoma - T-cell, B-cell, Null-cell
- (1) Diffuse Large B-cell Lymphoma - B-cell
- (2) Anaplastic Large Cell Lymphoma - T-cell, Null-cell, B-cell
HL → excellent
NHL → related to histo + stage
- HL ⇒ combination chemo +/- RadioT
- NHL ⇒ combination chemo, if CD20 pos → RITUXIMAB
{kind=link}
- Vascular stages (vasoconstriction)
- Platelet stage: Adhesion & Aggregation (platelet activation)
- Coagulation (plasmatic) stage (clot formation)
PRIMARY HEMOSTASIS ABNORMALITIES
- Vascular
- vasculitis
- hemorrhage
- Platelet
- ↓ Production in BM:
- BM failure (aplastic anemia)/ suppression / malignancy (leukemia, lymphoma)
- Infections
- ↑Turnover / Destruction
- ITP (or orther AI-diseases)due to Ab
- Drug induced (→ i.e. Heparin: HIT)
- DIC (+sepsis)
- HUS (hemolytic uremic syndrome)
- Pregnancy: preeclampsia + HELLP
- Distribution + Liver
- Distribution
- Splenomegaly (↑sequestration)
- vWD (2b)
- Liver disease (↓TPO production)
- Genetic (Glanzmann, Bernard-Soulier)
- Acquired:
- drugs: aspirin/clopidogrel
- CKD

other:

Preeclampsia:

HELLP: Acronym: Hemolysis Elevated Liver enzymes, Low Platelets
SECONDARY HEMOSTASIS ABNORMALITIES
- Intrinsic: Hemophilia A+B(+C)
- Extrinsic: F7 def. (rare)
- Both:
- Vit K def / Warfarin
- DIC

BLEEDING at different locations🤡
- spontaneously
- after minor trauma
- after trauma / surgery
→ check history!
- mucosa → nose, gum, menorrhagia
- Skin
- Petechia: 1-3mm
- Purpura: 3-10mm
- Echymosis: ≥1cm
- Hematoma
- subcutaneous
- muscles
- joints: hemarthrosis
- organ bleeding
- brain !




- Lymphadenopathy
- Hepato- + Splenomegaly
- Bleeding time
- Platelet count
- PPT, Quick(PT/INR), (Thrombintime)
- aPTT → instrinsic (+common)
- PT → extrinsic (+common)
- Fibrinogen/Thrombin time → Thrombin + Fibrinogen

{kind=link}
platelet function (+vascular) = PRIMARY HEMOSTASIS
(Øcoagulation)

🩸 BT | 🐦 aPTT | 🪂 PT | |
🍽️ Platelet (+vascular) | ↑ | ||
🤠 vW disease | ↑ | ↑ (F8) | n/↑ |
🅰️ Hemophilia A (F8) | ↑ | ||
🅱️ Hemophilia B (F9) | ↑ | ||
Heparin | ↑ | ||
Warfarin | ↑ (early) | ↑ (late) | |
DIC + Fibrinolysis | ↑ | ↑ | ↑ |
early: ↑PT
late: ↑APTT
Explanation:
Vitamin K promotes synthesis of coagulation factor of the extrinsic, intrinsic + common pathway:
(1) Factor II (2) Factor VII (3) Factor IX (4) Factor X
(5) Factor C (endog. anticoagulant) (6) Factor S (endog. anticoag.)
T
Ab against Platelets ⇒ 👾 Phagocytosis of platelets in spleen
⇒ ↓Lifespan & ↓Thrombocytes
⇒ ↑Megacaryocytes (compensatory BM activation)
- 🦠Recent viral infection (1-4w)
- 🩸Bleeding:
- acute onset generalized purpura
- classic (esp. muco-cutaneous)
- NO Leukemia-signs (LN, splenomegaly, leukopenia, anemia, fever)
CBC:
↓ thrombocytes 🤡 (<100k) ⇒ usually severe (<10-20k) Thrombocytopenia
Look for secondary causes:
(Could it be leukemia? Could it be Evan syndrome?
Abnormal ⇒ Indication for BM aspiration

(DDx malignancy) + Coombs test (Evan syndrome?)
Evan sy:

- SLE workup (ANA, anti-dsDNA)
- HIV workup
Normal physical exam (except bleeding syndrome)
PLUS
isolated (normal WBC,RBC)
PLUS
severe thrombocytopenia (<10k)
PLUS
Ø Tx was required OR Tx with IVIg
- Purpura
- Thrombos <20k (severe thrombocytopenia)
- Exclusion of DDx + Ømalignancy signs (ØHSM, ØLN)
- Thrombocytopenia DDx
- Distruction
- Distribution
- ↓ production (BM)
- Thrombopathies DDx
- ↑Megakaryocytes
⇒ see above
⇒ see above
20% (every 5th)
Criteria | Acute ITP | Chronic ITP |
M/F | 1/1 | 1/3 |
Age of Onset | <10 yrs | >10 yrs |
Onset Type | Acute | Insidious |
Infectious Prodrome | Yes (50-65%) | No |
Platelet Count | <20,000/mm³ | 30,000-70,000/mm³ |
Duration | <6 months | >6 months |

steroids #1: Predni +IVIG #2 Dexa + Rituximab
Cause:🧬 Genetic (autosomal transmission)
- too less
- To retarded
- None
Type | Description | Genetics | Prevalence |
1 | Partial quantitative deficiency of VWF | Autosomal dominant (recessive) | 70-80% |
2 | Qualitative deficiency of VWF | Autosomal dominant / recessive | 15-20% |
2A | Decreased platelet-dependent VWF function with selective deficiency of high-molecular-weight multimers | Autosomal dominant (recessive) | 10-12% |
2B | Increased affinity for platelet GPIb | Autosomal dominant | 3-5% |
2M | Decreased platelet-dependent VWF function with high-molecular-weight multimers present | Autosomal dominant | 1-2% |
2N | Markedly decreased binding of factor VIII to VWF | Autosomal recessive | 1-2% |
3 | Complete quantitative deficiency of VWF | Autosomal recessive | 1-3% |
- is it vWD?
- bleeding time: ↑ / n
- PTT: ↑ / n
- ↓Faktor 8
- Platelets normal (In Type 2B → ↓Platelets)
- vWF-specific studies (quantitative vs. qualitative)
- vWF-Amount ⇒ vWF-Antigen (immuno assey)
- vWF-Activity ⇒ Ristocetin cofactor test
- vWF-Structure ⇒ vWF multimers


Parameter | Hemophilia A | Hemophilia B | von Willebrand Disease |
Prevalence | 1/5000 boys | 1/25000 boys | 1/100-1/1000 (1/100 screening) |
Transmission | X-linked | X-linked | Autosomal dominant or recessive |
Factor Deficit | VIII | IX | von Willebrand and VIII |
Hemorrhage | Muscle, joints, surgery | Muscle, joints, surgery | Skin, mucous, surgery, menstruation |
APTT | ↑ | ↑ | ↑ or normal |
Bleeding Time | N | N | ↑/N |
Factor VIII | ↓ | N | ↓/N |
von Willebrand : Ag | N | N | ↓ |
von Willebrand : Activity | N | N | ↓ |
Factor IX | N | ↓ | N |
Ristocetin Aggregation | N | N | N/↓ |
Platelet Aggregation | N | N | N |
Treatment | DDAVP or factor VIII | Factor IX | DDAVP or factor VW |

*antifibrinolytics i.e caproic Acid
A → F8 deficiency (more common)
B → F9 deficiency

F <1% ⇒ severe
F 1-5% ⇒ moderate
F 5-25% ⇒ mild
Boy
(X-linked recessive transmission 📷)
{kind=link}
Severity | Trigger of bleeding | spontaneous bleeding? | specific bleeding |
mild >5% | signif. trauma | Ø | |
moderate <5% | trauma | (rare) | |
severe <1% | spont. / minor trauma/surgery | ✅ |
- ↑PTT + normal PT
- Specific assey for Faktor 8 / 9
Disorder: | 🩸 Bleeding | 🐦 aPTT | 🪂 PT (Quick) |
🍽️ Platelet (+vascular) | ↑ | ||
🤠 vW disease | ↑ | ↑ (F8) | |
🅰️ Hemophilia A (F8) | ↑ | ||
🅱️ Hemophilia B (F9) | ↑ | ||
Heparin | ↑ | ||
Warfarin | ↑ (early) | ↑ (late) | |
DIC + Fibrinolysis | ↑ | ↑ | ↑ |
Replacement therapy ⇒ Recombinant F8/9 concentrate (plasma)
1 IU = increases F8/9 with 2% in A & 1% in B per kg
IU/Dose = Weight x (necessary-patient level) x 0,5/1
hemorrhage/surgery | necessary level |
mild hemorrhage | (30-)40% |
mod/sev hemorrhage
small surgery | >50% |
life threatening hemorrage
complex surgery | 80-100% |
Type of Hemophilia | Factor Concentrate | 1U Increases Serum Level By | Dose Calculation Formula | Number of Doses/Day |
Hemophilia A | Factor VIII (FVIII) | 2% | (U FVIII)/dose = Weight (kg) x (necessary level - patient level) x 0.5 | 2 doses (every 12 hours) |
Hemophilia B | Factor IX (FIX) | 1% | (U FIX)/dose = Weight (kg) x (necessary serum level - patient level) x 1 | 1 dose (every 24 hours) |
⇒ prophylactic (lifelong) = 20-50 U/kg/dose; 1x/d; 3-4d/week
Add-on: (like vWD
if mild (+ mod.) hemophilia A ⇒ Desmopressin
Triggers the release of vWF endothelial cells→ ↑ factor VIII
if oral cavity hemorrhage/surgery ⇒ Antifibrinolytics
- Hematoma sequela
- hemophilic arthropathy
- Compressive hematomas
- Due to plasma Tx
- Infections
- HIV
- Hep B+C
- “Inhibitor” = Faktor 8/9 Ab



NO, contraindicated