





Genetics & Birth defects
- Trisomie 21 (down)
- Trisomie 18 (Edwards)
- Trisomie 13 (Pätau)
- 47, XXY (Klinefelter Syndrome)
- 45, X (Turner Syndrome)
- gonosomal and autosomal
- numerical and structural
1/600 children born alive! its the most common aberration
Age of the mother! 1/6 if the mother is 50 years old
Screening if ≥ 35
Mum age (years) | Trisomy 21 incidence |
20 | 1:1500 |
40 | 1:100 |
45 | 1:30 |
50 | 1:6 |
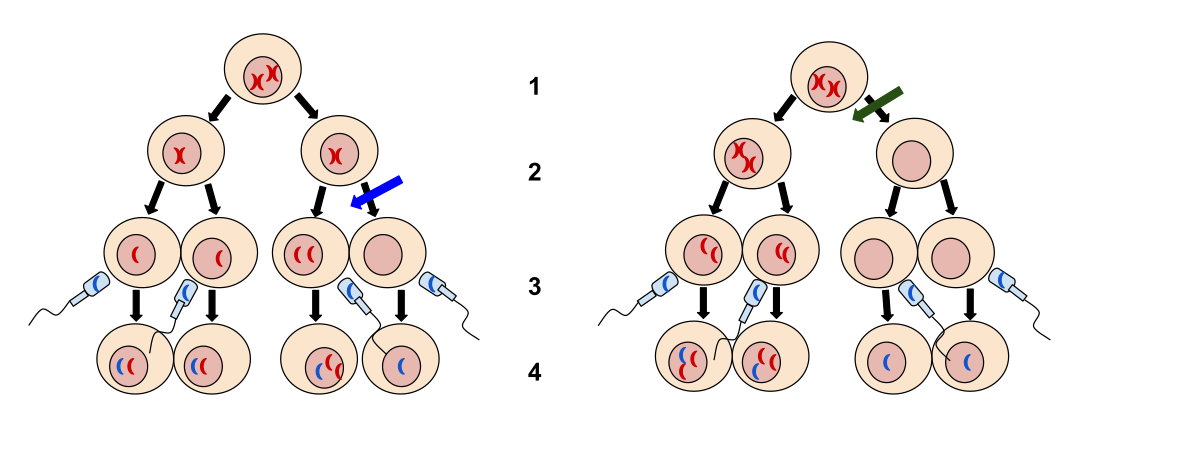
its not a hereditary dsease in the classical sense, as it results from a non disjunction during meiosis 📷
{kind=link}
(not during mitosis aka not mosaic pattern )
the information of the chromosome is tripled as well, but you can't see a third chromosome 21, as the chromosome is “ attached” (translocated” to another chromosome ( 14 most of the times) 46,XX,+21,t(21;14)
mosaic trisomie, only some cells have a trisomies ( 46, XX/47XX,+21)
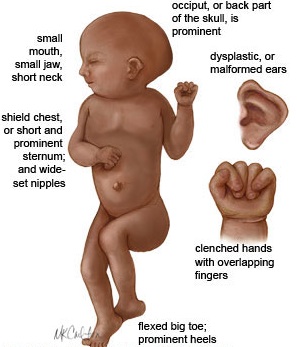
- outwards sloping eyelid axes (NO, YOU DONT SAY MONGOLOID ANYMORE!)
- Epikanthus
- Brushfield Spots

- Hypo/hyper-telorism
- Katarakt at early age
- small oral cavity
- big tounge (hanging out sometimes
- high palate
⇒ 📷
- 💀Brachyzephalus
- 👃hypoplastic nosebones
- 👂anormal ears (tiny, round, adherent earlobes)
50% of cases Heartdefect (ASD; VSD, AVSD, Fallot)
- duodenal atresia/stenosis + other intestinal atresias,
- pancreas anulare (Its circular )
- (also tracheal stenosis)

- pyloric stenosis, malrotation
- Hernias: inguinal+umbilical
- morbus hirschsprug
- megacolon
- rectal prolaps
- reduced fertility in males
- hypogonadism
- 🌱increased risk for leukemia
- 🦋hypothyreosis
- 👂reduced hearing due to repetitive otitis media
- 🥐 - hydrophrosis, polycystic kidney, hypoplasia
- 📈 ↓growth velocity + height
- 🧠IQ around 50, but in mosaic forms, the intelligence can be only little reduced to normal
- Chorionvillibiopsy (very dangerous actually, up to 1% risk, that the child dies afterwards (or at least the number used to be so high))
- Amniocentesis
- chromosomalabberation can be detected in fetal DNA that can be found in the mothers blood
- very often false positiv
- hint that the child has trisomies 21: Free estriol ↓, AFP (alpha fetoprotein) ↓, ß HCG ↑ (at week 15)
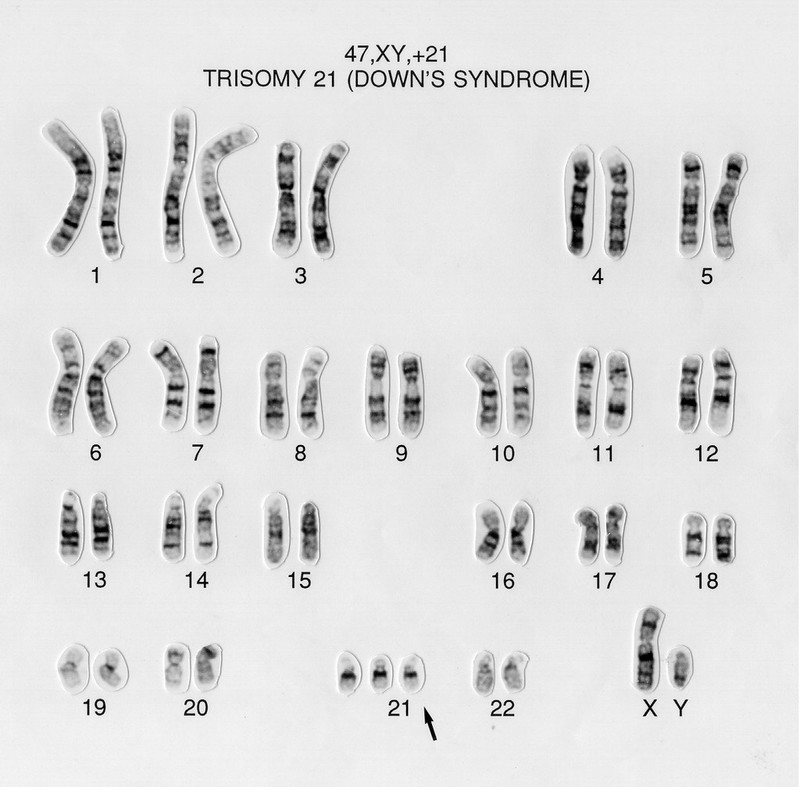
chromosomal analysis hehe (karyotyping) 📷
{kind=link}
NOtiiiing coz its genetic…
No basically you treat the complex symptoms of the child by doing heart surgery, GI surgery, early targeted support
lifetime is reduced due to the organdysmorphia and increased leukemia risk
most patient develop 📝Alzheimer symptoms until they turn 40
♀:47,XX,+18; ♂: 47,XY,+18
2nd most common trisomy (after 21)
associated with age of mother
3x more common in females
⇒ 📷
{kind=link}
- 💀
- Prominent Occiput
- Microcephaly + micrognathia
- 👂Low set ears
- 🐰Cleft Lip + palate (Lippen-kiefer-Gaumen-Spalte)
- ✊Overlapping fingers in clinched hands
- 🎸Rocker- bottom feet

- 🫀Congenital heart defects
- 💩Gastroentestinal anomalies
- 🧱 Omphalocele 📷
- 🌯 oesophageal atresia (blind ended oesophagus)
- 🥐 Kidneymalformations (Horseshoe kidneys🐎)
- 🦠 Frequent infections

→ Increased risk for developing hepato- and nephroblastoma
- 🧠 intellectual disability - 🤪 SEVERE RETARDATION
- 📈failure to thrive (weight increase) + growth retardation + psychomotor retardation
- 🍇 puberty → sexual characteristics appear later
US: Nuchal translucency
Prenatal sonogram: polyhydramnios
Serum test: HCG, PAPP-A, AFK, Inhibin
Karyotyping (prenatal = amniocentesis; postnatal = blood)
☠️ 90% die in the 1st year
cry of the cat 🐱
deletion of a small part of chromosome 5 , (46, XX, 5p-; 46, XY, 5p-)
- 🐱 cry sounds like a cat
- 🧠 severe mental retardation 🤪
- 👶 small head with unusual facial features ⇒ 📷
45, X0 (monosomy X)
Only female phenotype, caused by non-disjunction during meiosis (45 X0) or non disjunction during mitosis (45 X0, 46 XX → mosaic form) ⇒ like trisomy
Faaaaalse 📷
{kind=link}
T 📷
{kind=link}
chromosmal nondisjunction → X Chromosome monosomy/mosaic → lack of information from the second X chromosome → impaired ovarian development → malfunctioning gonads, have connective tissue instead of normal germ cells (so they are infertile) → leads to estrogene + progesterone deficiencies
cystic changes, grotesque anomalies, often they are miscarriaged
- Lymphedema ( Cystic hygroma and lymphedema of hands and feet) →🕸 neck webbing
- 💀dysmorphism
- 🌶aortic coarctation
juvenile phenotype ⇒ see below
- 🦴
- osteoporosis with pathological fractures
- 🧰broad chest with wide space nipples
- 💪 weird angle of the elbow (Cubitus valgus)
- 🔽 and they are very shoooort, like super short
🌶️AORTAAAAAAAAAA
- bicuspid aortic valve
- aortic
- Coarctation of the aorta
- aortic dissection and rupture
- hypertension

🥐 The horseshoe kidney again 🐎
+🥚ovarial dysunction (ovarial streak)
They have a perfectly normal IQ ✅
(also normal speech, spatial orientation might be impaired)
T, until the age of 3 ⇒ afterwards the growth rate decreases
122-147 cm
its very delayed
no, the girls have a
- primary amenorrhea,
- underdeveloped breast,
- almost no pubic hair
they can’t , as they don’t have their own eggcells, but as they have an uterus, the can become moms by IVF (with a donor egg) funfact: in Germany you can donate sperm, but no eggcells 🙃🙃🙃
THEY ARE SHOOOORT! ( in case u forgot)
🦋
Hashimoto thyroiditis ( 🐢🍁 hash smoking turtle 🐢🍁)
yes, diabetes, but type 2
🍬
Estrogen replacement therapy or estro-progestative therapy
With growth hormone (GH) administration!
at age 4-5
(Aka after they stop to grow normally)
anabolic (androgenic) steroids 💪🏼 at 10 years

Streak ovaries
short statue
neck webbing
lymphedema
its a numerical GONOSOMAL abnormality 47, XXY
male hypogonadism
nondisjunction of the X chromosome
- ↑ FSH + LH
- ↓ Testosterone , ↑ aromatase , ↑ estrogen
- 🍈 Gynecomastia (increased risk for breast cancer!)
- 🧔🏻 sparse body hair
- 🥜
- small testes
- infertility
→ Due to the second X chomosome, the production of tetsosterone is significantly lower —> leading to a more “female” phenotype
- 🧠 cognitive: moderate mental retardation, learning difficulties
- 🤸🏼 behavior abnormalties (immature, introverted, antisocial etc)
- ⬆️ tall staute
- ⚖️ excess weight
- normal bone age, but abnormailties of bones sometimes (increased risk for osteoporosis)
Testosterone subsitution after the age of 10-11 years
mastectomy and orchidopexy
{kind=link}
{kind=link}
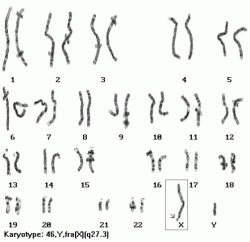
females have another X, can cause premature ovarian insufficiency in females (menopause before 40)
55-200 repeats, only mild/non symptomes
{kind=link}
- 🧠 Intellectual disability (varible: mild-severe)
- 🧩 5% of cases assoc. with autism (genetically caused)
- 🤸🏼 behavioral abnormalities
- 🧤 stereotypic movements (hand-flapping)
- 😰 social anxiety
a clinically significant block of a metabolic pathway due to a single gene defect
often enzymes can be subsitutet and long term consequences can be avoided
Screeeeeening plz
T and F , 🤡taken as a group, they are fairly common, but the single conditions are rather rare
- accumulation of toxic substrates (generalized/localized)
- abnormal metabolism with formation of toxic metabolites
- lack of production and normal metabolites
- acute onset of symptomes
- “Sepsis” in infants
- Vomiting, esp. after changing of diet
- failure to thrive, loss of appetite
- hiccups
- jaundice, hepatomegaly, reye like syndrome
- unusual odors of urin/body
- unexpl. haemorrage
- psychomotor delay, development regression
- apnea, resp. distress, hypervent.
- Quick resp to IV fluid
- enzyme replacement therapy
- pharmacologic doeses of enzyme cofactor
- cause correction
- gene therapy
- liver/bonemarrow transplant
- dietary exlusion
- removal of substrate /toxic metabolites
- dietary supplements with not synthesized metabolites
- stimulations of alternative pathways
- discontinuation of oral diet
- IV/PO nutrition with glucose and electrolytes
- Treatment of complications: shock; hypoglycemia/acidosis….
- Vitamine subistution
- CLEAN UP
- detoxyfication for urea cycle defects
- peritoneal dialysis
- haemodialysis
you cant lel, its a genetic disease🤡
but you can do genetic counseling, prenatal diagnostic and newborn screening (not diagnostic)
- congenital hypothyroidism
- phenylketonuria,
- galactosemia (classic)
- galactokinase deficit
- maple syrup urine disease
- homocystinuria
- biotinidase deficiency
- congenital adrenal hyperplasia
- sickle cell disease
⇒ 📷
{kind=link}
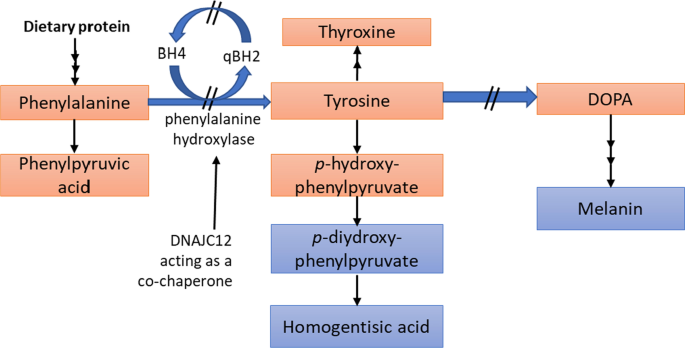
Due to the mutation of the Phenylalanine hydroxylase enzyme the aminoacid phenylalanine cannot be metabolized to tyrosine → Phenylalanine accumulates and tyrosin levels drop
- autosomal recessive metabolic disorder
- Phenylalanine hydroxylase gene is located on choromosome 12 (bands 12q22-q24.1)
- most patients are heterozygot for two dofferent mutations
tyrosine, dopamine, norepinephrine and epinephrine, melanine
Phenylalanin uses the same transporters to get across the blood-brain barrier as Tyrosine and Tryptophane. When Phenylalanin levels rise, the transporters are all occupied, so tyrosine and tryptophane cant get across the barrier
phenylalanin levels rise in the body fluids and CNS
{kind=link}
Tyrosine is needed for norepinephrine and dopamine and tryptophan for serotonin, if the aminoacids cant get across, all of the transmitters are in reduced concentrations in the brain, leading to abnormal brain development and intellectual disability
melanin deficency, leading to light sensitivity
it is metabolized into Phenylketones and excretet in the urine (That why it is calles PKU),
the ketones dont play an role in the pathogenesis
>20mg/dl in the serum
asymptomatic 📷
{kind=link}
🧠
- 🤪Mental retardation (gradually)
- 👇🕺hyperactivity, athetosis (if not treated)

- 🗽UMN signs: hypertonia, exaggerated reflexes (hyperreflexia)
- ⚡️EEG changes (50%)
- Seizures
- 🤮Vomiting (DDx: Pyloric stenosis)
- 💪🏼Skin: Sebheroic dermatitis, eczema
- 🍎fruity odor (acetonic odor) → mouth and urine
- 💀Microcephaly (if maternal PKU) with prominent jaw + spaced teeth + growth retardation
mild deficency of the Phenylalanin hydroxylase or a lack/deficency of the cofactor (BH4)

They might stay asymptomatic, but progressive brain damage occurs anyways with age, the dont smell, because they dont excrete phenyl-ketones
genetic probes from chorionzottenbiopsie (chorion villi biopsy)
24-48 h after birth
a quantative determination of plasma phenylalanin wil be done, so another test basically
and cofactor deficency needs to be ruled out
quantitative determination of phenyl alanine via flurometic of mass spectrometry from a drop of blood taken from the cilds heel after protein consumption 📷
{kind=link}
Phenylalanine ketone detection → detects children in countries without screening
phenylalanine free/ low food (no protein rich foods) ASAP
>6mg/dl
phenylalanin is an essential aminoacid (lack leads to side effects like lethargy, failure to thrive, anorexia, anemia, rash, diarrhea, and death)! but in PKU patients Tyrosine becomes an essential aminoacid —> substitution
oral tetrahydrobiopterin (BH4) might lower the levels (in addition to low phenylalanine diet)
→ cofactor for phenylalanine hydroxylase
Palynziq: Enzyme applicated every day subcutaneously and metabolizes Phenylalanin in the blood
its part of lactose (galactose+ glucose = lactose)
⇒ 📷
{kind=link}
under the umbrella term galactosemia one understands an autosomal recessive genetic disorder, that affects the ability to metabolize galactose → enzymatic deficency
⇒ 📷
- Type I:
- deficency in galactose-1-phosphate uridylyltransferase (GALT) = classic
- Type II:
- deficency in galactokinase (GALK)
- Type III:
- deficency in UDP-galactose-4-epimerase (GALE)
BUILDUP of GAL1-Phosphate → hepatotoxic, failure to thrive (lethargy, n/v, diarrhea, poor feeding), sepsis
Galactose-1-Phosphate inhibits pyrophosphorylase that leads to decreased UDP glucose and UDP galactose levels UDP galactose is importaint in neonates, as it is importaint for:
- bilirubin conjugation
- brain myelin formation
- post transtlational processing of FSH, membrane proteins
Galactose 1 phosphat uridyl transferase
= GALT
Converts galactose 1 phosphate to UDP-galactose, UDP galactise gets converted to UDP glucose → Glucose 1 P
in severe cases yes, in partial defects ist asymptomatic
accumulation of toxic substances
- 🤮💩Vomiting, diarrhea
- Poor feeding
- Failure to thrive
- 🍑Liver failure:
- 🌼Jaundice + hepatomegaly
- ↓🍬 Hypoglycaemia
- 🥐 Renal tubular dysfunction ⇒ LOSS OF:
- bicarbs → 🧪acidosis
- 🚵 albuminuria + 🍬glycosuria
- 👁️Cataract

- 🧫 ↑ Risk of E.coli sepsis (esp. in neonates) (if a child has unexplained E coli infection think of galactosemia!!)
- 🙋♂️ older children:
- 🧠Cognitive impairment → learning problems
- 🥚premature ovarian failure in girls
Galactose ↑ (blood + urine) Galactitol ↑ (blood + urine) Galactose-1-P ↑ (blood)
Galactokinase, Converts galactose to galactose-1-phosphate
- Accumulation of galactitol in tissues (alternative pathway to metabolize galactose)
- Galactose and galactitol are present in blood and urine
- Cataracts👁️, white pupill reflex
- Pseudotumor cerebrii
- ØLiver disfunction
UDP-galactose-4-epimerase, converts UDP-galactose to UDP-glucose
its mostly asymptomatic, but esp UDP galactose accumulates
usually asymptomatic
if severe TRIAD: ⇒ Type 1 + Hypotonia + sensorineural deafness
⇒ 📷
Galactose, Galactose 1-P, Galactitol, UDP-Galactose
You want to check the Urin and blood
- Plasma:
- ↑🥛 Galactose
- 🧪 metabolic acidosis
- ↓🍬hypoglycemia
- RBC
- 1️⃣↑ Galactose 1 phosphate
- ↓GALT

- Galactitol (in type I)
- Albuminuria
- Positive/negative Clinistix
→ Test detects reducing substances like galactose (negative does NOT exclude galactosemia, can be false positive due to renal dysfuntion)
other cases of liverfailure:
- hepatitis
- biliary obstruction
- fructose intoleranca
- tyrosinemia type I
- Lactose free diet ⇒ soy-based diet instead
- Education of parents and older children about the galactose content of foods
🍦Calcium + 🦴Vit D suppl bc no milk
🥚 ovarian failure Tx
FOLLOW UP regular:
- 🧠language evaluation at 3
- 👁️eye examination (cataract)
if diet ist retricted from 7 days on, heapatotoxic manifestations will regress, good prognosis
delayed therapy → mental retardation and reduced growth
detection of parental genetic carraiage of the mutation
prenatal diagnosis
neonatal screening
an enzyme defect that leads to problems in the glycogen synthesis or the breakdown
🍑 + 💪 + 🫀
liver and muscles + heart
its an autosomal recessive disease, that is caused by mutations in genes coding for enzymes involved in the glycogen metabolism in the liver/muscle
classically 6, but actually 13 are discribed
in muscle and/or liver GSD
TYPE 2, 3, 4, 5
- 💪↓glycogen breakdown for muscle activity
- Easy fatiguability, exercise intolerance
- Type V: Second wind phenomenon - fatigue disappear after period of activity
- Cramps
- Rhabdomyolysis → myoglobinuria
- ❤️Cardiac involvement (II and III)
- ↕️ cardiomegaly (HCM)
- 🎶conduction defects
(glycogen = source for intensive muscle activity)
Ø glycogen breakdown → symptoms after short period of intense exercises DDx defecs of lipid metabolism → symptoms after prolonged exercise
⇒ MUCLE EXHAUSTING / DAMAGE:


TYPE 1, 3, 4 (6)
- 🍑Hepatomegalia → distended abdomen
- 🍬 Hypoglycemia + Ketosis (↓glycogen breakdown)
- ⇒ Signs of hypoglycemia in infants:
- 🧠seizure + irritability + hypotonia,
- 🫐cyanosis
- ↓🍼poor feeding,
⇒ only in I + III + Ø in type IV
Type 4 usually Ø hypoglycemia (unless liver failure). T1 least ketosis, but worst hypoglycemia episodes T3 mild glycemia, present later than T1
📈failure to thrive
🐼anemia (1)
🧈hyperlipidemia (1+3)
- detection of mutation in leukozytes (no biopsy necessary!)
- enzymes in the affected tissue (when DNA mutation cannot be detected due to lack infrastructure)
- metabolic load/exercise test
avoid hypoglycemia,maintain blood glucose at 80-90 mg/dl
continuous meals during the day and gastric feeding at night Restriction of galactose and fructose (cannot be converted into glucose)
(type II) substitution therapy (apha glucosidase)
a deficency of specific enzymes, that lead to the accumulation of of abnormal substances that are usually degraded within lysosomes, resulting in cell damage and death (not nice:( the have a progressive course
(i.e. gaucher + MPS)
lysosomal membran syntheses
dysfuntion of lysosomal transport proteins
the gene for beta Glucocerebrosidase (GBA) is mutated, leading to a ↓ β-glucocerebrosidase level,
its an autosomal recessive disease of the GBA gene
glucocerebrosides can’t be broken down and accumulates Its a sphinpgolipid 🍔 found in cellmambranes → cant be broken down in the lysosomes of the macrophages, leading to Gaucher cells, that accumulate in the brain, liver, spleen and bone marrrow ⇒ gaucher cells release lysosomal enzymes + cytokines ⇒ immune response ⇒ scarring
- 🦴 (not in type 2)
- BM fibrosis ⇒ ↓blood cell productiction: 🐼anemia + ⚪ leukopenia
- bone 💥PAIN
- Bone infarction ⇒ ☠️ avascular necrosis
- Osteoporosis
- 📈 Growth delays
- 🍑 🍠 HSM
- 🔵 thrombocytopenia ⇒ pancytopenia (w BM fibrosis)
- 🫁 Pulmonary manifestations
- Type I: non-neuronopathic
- Type II: acute neuronopathic
- Type III: chronic neuronopathic
🧠
not as importaint, but to be complete
main difference:
Type 1: NO brain involvement (= most common 🥇)
Type 2: acute 🧠neurodegeneration + Type 1 , →☠️ death within 1st year
Type 3: like Type 2🧠 but gradual onset
Characteristic | Type 1 (Chronic Adult Non-neuronopathic) | Type 2 (Acute Infantile Neuronopathic) | Type 3 (Subacute/Chronic Juvenile Neuronopathic) |
Demographic Group | Ashkenazi Jewish | --- | Norrbottian Swedish |
Age at Onset | Any (infants to adulthood) | Infancy (3-6 months) | Childhood |
Skeletal Disease | Absent to severe | Absent | Moderate to severe |
Hepatomegaly | Mild to extreme | Moderate | Mild to extreme |
Splenomegaly | Mild to extreme | Moderate | Mild to extreme |
CNS Disease | Absent | Significant | Mild to significant (increasing with age) |
Life Span | 6 yrs to more than 80 years (normal with enzyme replacement) | <3 yrs | 2-60 years |
🦴 + HSM:
- 🦴bone-pain
- unexplained 🍑🍠hepatosplenomegaly
- easy 🔵bruising (thrombocytopenia)
- beta glucocerebrosidase activity levels in WBC
- 🧬-testing: mutation identification
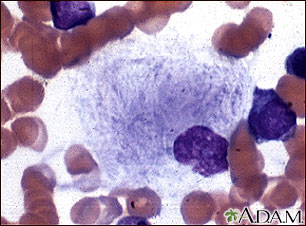
Gaucher cells (macrophages + inclusion) ⇒ “wrinkled paper appearance” (intra-cellular inclusions of gluco-cerebro-sides) 📷
{kind=link}
- enzyme replacement therapy (glucocerebrosidase)
- most things can be reversed/stabelized (liver, spleen, bone)
- but the 🧠neurologic progression can Ø be stopped
- substrate reduction therapy ⇒ blocks cermaide glucosyltransferase inhibitor →↓glucocerbroside synthesis
its CURATIVE but high morbidity and mortality
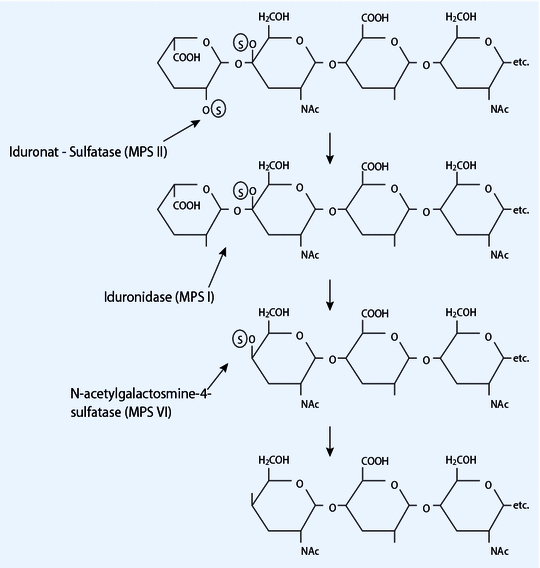
lysosomal enzyme mutation: alpha-L-iduronidase 📷→ impaired breakdown of mucopolysaccharides (heparin-sulfate + dermatan sulfate) → no breakdown ⇒ build-up of mucopolysachhardies synonym mucopolysachhardies = glycosaminoglycans
{kind=link}
{kind=link}
Hurler
Scheie
Hurler-Scheie (lol)
* there are a lot more, and these are only the subtypes of MPS I, but its the only one mentioned in the book)
Characteristic | Hurler Syndrome | Scheie Syndrome | Hurler-Scheie Syndrome |
Severity | Severe | Mild | Moderate |
Onset | 6-24 months | >5 yrs (10-20 yrs) | 3-8 yrs |
Impaired Mental Development | + ! | - | ± |
Corneal Clouding | + | + | + |
Hepatosplenomegaly | + | - / (+) | + |
Skeletal Defects | + | - | + |
Coarse Facial Features | + | (+) | ± |
Cardiac Disease | + | - ! | + |
Motor Weakness | + ! | - | - |
Hernia | + | - | - |
Stiff Joints | + | + | - ! |
Short Stature | + | - | - |
Life Span | <10-14 yrs | adulthood | adulthood |
↑Urinary Mucopolysachharides (dermathan sulfate + heparin sulfate) PLUS suggestive clinic + radiological signs
↓
Confirmation: alpha-L-iduronidase enzyme assay in WBC
- hematopoetic stem cell tranplantation (< 2 yrs)
- enzyme replacement: Alpha-L-Iduronidase (before + after transplant)
bot things are you born with, but congenital means, it is a condition present at birth regardless of its cause ( can be due to infections, teratogenic factors, epigenetic…) and genetic means it has a genetic cause in terms of mutation
T
- preconceptional
- genetic disease
- postconceptional
- congenital (not-genetic)
- environmental factors
chemical factors | |
drug | antibiotics
Anticoagulants
Antiepileptics
Psychotropes |
toxines | Alcohol
Cocaine
Pollutants
Tobacco |
physical | Ionizing radiation
Excessive heat
Traums |
infectious | Syphilis
TORCH:
Toxoplasmosis
Other infections (syphilis)
Rubella
Cytomegalo
Herpes simplex 2 |
mechanical | amniotic disease |
maternal | diabetes, anemia, fetal blood dyscrasia, placental disease |
nutritional | hypovitaminosis
Protein deficency |
endocrine | diabetes |
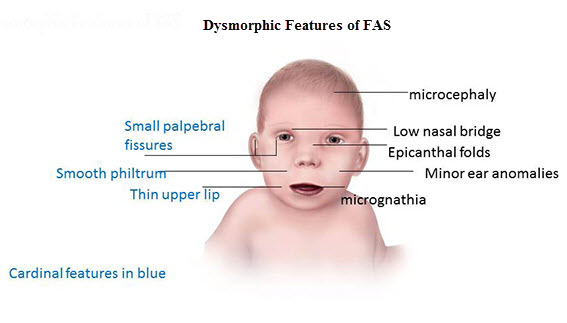
11% risk if mother consumes more than 25-50g/day of alcohol
⇒ 📷
{kind=link}
- microcephaly💀
- epicanthus👁
- 👄
- Thin upper lip
- indistinguishably filtrum
- ❤️Congenital heart defect
- 🤪mental retardation
- 🕺hyperactivty
- SMALL: ⚖️low birth weight + 📈growth delay
Congenital ❤️ defect treatment,
supportive
TOXOPLASMOSIS OTHER INFECTIONS (SYPHYLIS, PARVOVIRUS B19) RUBELLA CYTOMEGALOVIRUS HERPES SIMPLEX VIRUS
yes exaclty, toxoplasma gondii, trandmitted by cats 📷 and pigenos in unhygenic settings
{kind=link}
transplacental 📷
{kind=link}
- 📛 septicemic form
- Severe, can be lethal
- 🧠encephalomyeletic form
- affects CNS
- 👇 oligosymptomatic form
- ⏰latent form (55%)
- Classic triad
- 👁Chorioretinitis
- 🧠
- Diffuse intracranial Calcification
- Hydrocephalus
- sytemic disease → encephalitis
- most die
- those who surive only with severe neurological disorders (and rinitis pigemntosa)
The 4 Cs of congenital toxoplasmosis: Cerebral calcifications, Chorioretinitis, hydroCephalus, and Convulsions.
- normal at birth
- Early onset of the disease
- microcephaly, neurological disorders
- child asymptomatic carrier
- during adolescence deafness and blindness → retinitis pigmentosa

🧠 epilepsy + intellectual disability
👁 Visual disturbances
👂- sensorineural hearing loss
- clinical diagnosis
- laboratory identification of IgM against T. gondii
- Or PCR
- specific anti-toxoplasma IgM and IgG AB in maternal serum
- Ophtha exam
pirimethamine, sulfadiazine, spiramycin, prednisone, and folic acid (don’t remember)
→ 1y of therapy
→ mother shoul be treated as well
treponema pallidum 📷
{kind=link}
during birth 📷
{kind=link}
- early severe
- common forms
- early congenital syphilis
- late congential syphillis
- latent asymptomatic (only sero-positive)
MULTIORGAN INFLAMMATION
- 🍑liver
- 🫁lung
- 🥐kidney
- LIKE THE OTHERS (see later)
- 💪 👃 skin + nose
- 🧠👁️ brain + eye

- onset at birth
- 💪skin disorders (exanthema, pemphigus)
- 👃🩸bloody syphilitic rhinorrhea
- 🦴syphilitic periostits+ osteochondritis

clinic +
- Serology: treponema test + non-treponema test
clinic+
- serology IgM (igG can cross the placenta)
- X-ray (bone involvement)
- Penicillin G → LOW DOSE ❗ (⇒ to avoid HERXHEIMER REACTION!)
Jarisch-Herxheimer reaction is a reaction to endotoxin-like products released by the death of harmful microorganisms (mainly treponema pallidum) during antibiotic treatment.
AB therapy → lysis of bacterial cell membranes → release into the bloodstream of bacterial toxins →systemic inflammatory response. ⇒ can cause a significant drop in blood pressure, acute end-organ injury, multi-organ failure (sepsis)
- Tx of maternal syphilis ⇒ can prevent congenital syphilis if AB started before 16th week
T
point in time of infection
- 85% malformation during first 8 weeks
- 16-23% if infection during 13-26 weeks
- 32% during 3rd month
tranplacental
- ❤️ Congenital heart disease: patent ductus arteriosus, septal defects.
- 👁️ Eye lesions: microphthalmia, cataract, glaucoma, chorioretinitis.
- 👂 Hearing impairment: deafness.
CCC-Triad of congenital rubella syndrome: Cataracts, Cochlear defects, Cardiac defect
- 💀microcephalia
- 🫀myocarditis
- 🍑hepatosplenomegly, jaundice
- 🦴BM: Amegakaryocytic thrombocytopenic purpura
- 🧠
- autism/mental retardation
- epilepsy
the virus can stay latent and cause encephalitis during adolesence
(asymptomatic from birth)
- identify during pregnancy!
- clinical pic
- IgM (IgG NO VALUE! might be passed through placenta)
Treat each disorder
💉 Vaccinate Woman against rubella, that want to get pregnant!
or asymptomatic🙅♂️
influenza like 📸
-w/ mild parotid swelling + low grade fever
generalized, localized and latent
- 🅱️ -symptoms
- fever
- Weakness
- 💀microcephaly
- ❤️CHD (congenital heart disease)
- 🧠CNS abnormalities
- 👂deafness
- 🍑🍠HSM
- 🦴BM:
- thrombocytopenic purpura
- anemia
⇒ usually severe course (lethal or neuro sequelae)
⚡neuro-pain
OR
🦴thrombocytopenic purpura
OR
🍑neonatal hepatitis
asymptomatic ⇒ only sero-positive
Anti-CMV IgM determination
clinical presentation +
- Anti CMV IgM
- virus in the urine
- localized → symptomatic Tx
- Anti-🦠: Gancyclovir (like Aciclovir)
sounds like Samwise Gamgee 📷
{kind=link}
- 🧠Cerebral calcifications
- 👁Chorioretinitis
- 🎈HydrCephalus
- ⚡️Convulsion
- treated with primethamine
- multiorgan inflammation
- Hutchinson triad:
- 🦈Hutchinson’s teeth - aka shark teeth
- 👁️interstitial keratitis (Inflammation of the cornea)
- 🦻 Deafness due to auditory nerve disease
- 🦴Clutton sign
- Treatment: Penicillin G (herxheimer!)
classic triad:
- ❤️ Congenital heart disease: patent ductus arteriosus, septal defects.
- 👁️ Eye lesions: microphthalmia, cataract, glaucoma, chorioretinitis.
- 👂 Hearing impairment: deafness.
3C: Cataract, Cochlear defects, Cardiac abnormalities
Treatment: symptomatic
- 🅱️ -symptoms
- fever
- Weakness
- 💀microcephaly
- ❤️CHD
- 🧠CNS abnormalities
- 👂deafness
- 🍑🍠HSM
- 🦴BM:
- thrombocytopenic purpura
- anemia
Treatment: symptomatic and ganciclovier
- hypergylcemia or hypoglycemia
- vasculopathy + placental dystrophy→fetal hypoxia
- Maternal hyperglycemia causes fetal hyperinsulinemia → macrosomia (coz its anabolic)
- ketose (maternal)
- intrauterine death ☠️
- 🍬Glucose effects
- metabolic abnomralities at birth
- seizures 🧠⚡
- macrosomia → ↑risk for birth trauma
- 💀❤️👁️🦴structural abnormailties (CNS, skeletal, facial, eye, CV…)

its a high risk pregnancy (control every 4-8w)
⇒ close diabetic control
in children → pathogenetic tx + surgery